

AMAS: a fast tool for alignment manipulation and computing of summary statistics
- PMID: 26835189
- PMCID: PMC4734057
- DOI: 10.7717/peerj.1660
AMAS: a fast tool for alignment manipulation and computing of summary statistics
Abstract
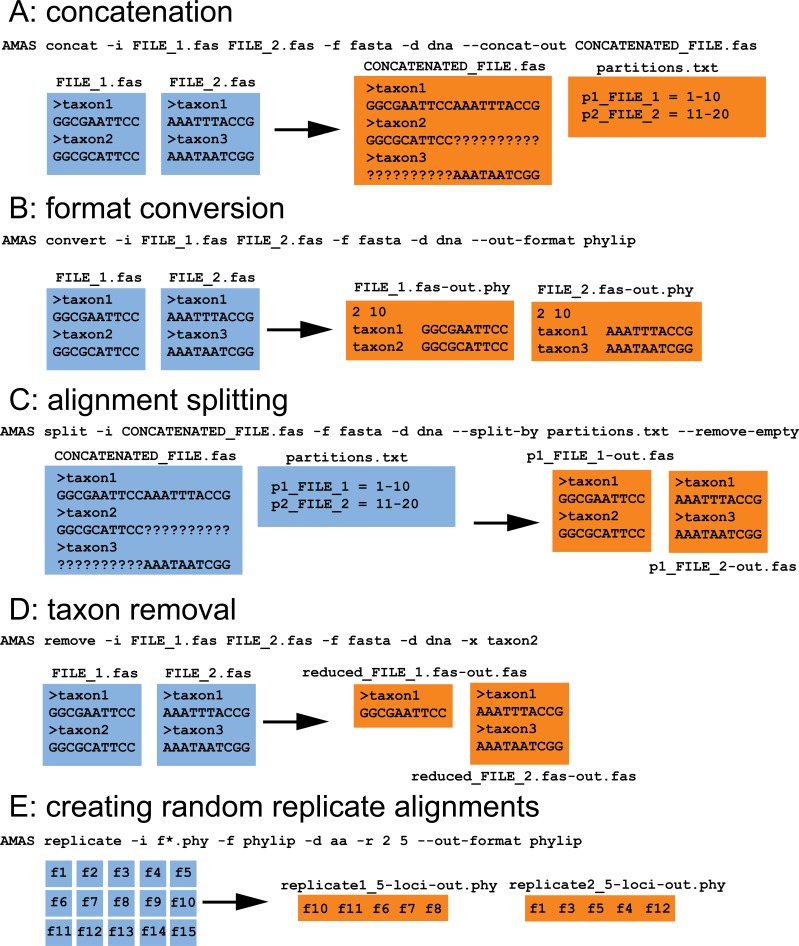
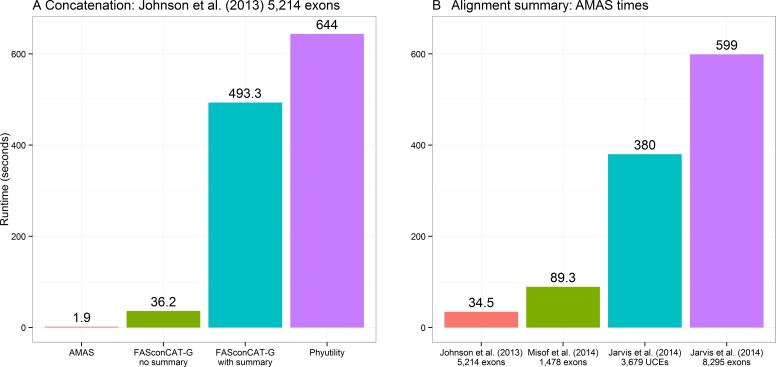
The amount of data used in phylogenetics has grown explosively in the recent years and many phylogenies are inferred with hundreds or even thousands of loci and many taxa. These modern phylogenomic studies often entail separate analyses of each of the loci in addition to multiple analyses of subsets of genes or concatenated sequences. Computationally efficient tools for handling and computing properties of thousands of single-locus or large concatenated alignments are needed. Here I present AMAS (Alignment Manipulation And Summary), a tool that can be used either as a stand-alone command-line utility or as a Python package. AMAS works on amino acid and nucleotide alignments and combines capabilities of sequence manipulation with a function that calculates basic statistics. The manipulation functions include conversions among popular formats, concatenation, extracting sites and splitting according to a pre-defined partitioning scheme, creation of replicate data sets, and removal of taxa. The statistics calculated include the number of taxa, alignment length, total count of matrix cells, overall number of undetermined characters, percent of missing data, AT and GC contents (for DNA alignments), count and proportion of variable sites, count and proportion of parsimony informative sites, and counts of all characters relevant for a nucleotide or amino acid alphabet. AMAS is particularly suitable for very large alignments with hundreds of taxa and thousands of loci. It is computationally efficient, utilizes parallel processing, and performs better at concatenation than other popular tools. AMAS is a Python 3 program that relies solely on Python's core modules and needs no additional dependencies. AMAS source code and manual can be downloaded from http://github.com/marekborowiec/AMAS/ under GNU General Public License.
Keywords: Alignment properties; Bioinformatics; Concatenation; Phylogenetics; Phylogenomics.
Conflict of interest statement
The author declares there is no competing interests.
Figures
References
-
- Cock PJA, Antao T, Chang JT, Chapman BA, Cox CJ, Dalke A, Friedberg I, Hamelryck T, Kauff F, Wilczynski B, De Hoon MJL. Biopython: freely available python tools for computational molecular biology and bioinformatics. Bioinformatics. 2009;25:1422–1423. doi: 10.1093/bioinformatics/btp163. - DOI - PMC - PubMed
-
- Jarvis ED, Mirarab S, Aberer AJ, Li B, Houde P, Li C, Ho SYW, Faircloth BC, Nabholz B, Howard JT, Suh A, Weber CC, Da Fonseca RR, Alfaro-Núñez A, Narula N, Liu L, Burt D, Ellegren H, Edwards SV, Stamatakis A, Mindell DP, Cracraft J, Braun EL, Warnow T, Jun W, Gilbert MTP, Zhang G, The Avian Phylogenomics Consortium Phylogenomic analyses data of the avian phylogenomics project. GigaScience. 2014;4:4. doi: 10.1186/s13742-014-0038-1. - DOI - PMC - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
