

Phenylketonuria: translating research into novel therapies
- PMID: 26835324
- PMCID: PMC4729111
- DOI: 10.3978/j.issn.2224-4336.2014.01.01
Phenylketonuria: translating research into novel therapies
Abstract
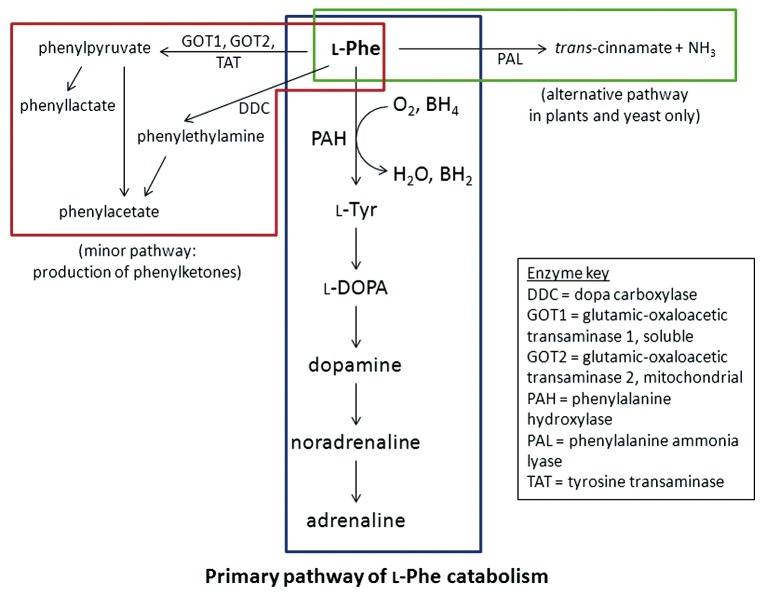
Phenylketonuria (PKU) is an inborn error of metabolism of the amino acid phenylalanine. It is an autosomal recessive disorder with a rate of incidence of 1 in 10,000 in Caucasian populations. Mutations in the phenylalanine hydroxylase (PAH) gene are the major cause of PKU, due to the loss of the catalytic activity of the enzyme product PAH. Newborn screening for PKU allows early intervention, avoiding irreparable neurological damage and intellectual disability that would arise from untreated PKU. The current primary treatment of PKU is the limitation of dietary protein intake, which in the long term may be associated with poor compliance in some cases and other health problems due to malnutrition. The only alternative therapy currently approved is the supplementation of BH4, the requisite co-factor of PAH, in the orally-available form of sapropterin dihydrochloride. This treatment is not universally available, and is only effective for a proportion (estimated 30%) of PKU patients. Research into novel therapies for PKU has taken many different approaches to address the lack of PAH activity at the core of this disorder: enzyme replacement via virus-mediated gene transfer, transplantation of donor liver and recombinant PAH protein, enzyme substitution using phenylalanine ammonia lyase (PAL) to provide an alternative pathway for the metabolism of phenylalanine, and restoration of native PAH activity using chemical chaperones and nonsense read-through agents. It is hoped that continuing efforts into these studies will translate into a significant improvement in the physical outcome, as well as quality of life, for patients with PKU.
Keywords: Phenylalanine hydroxylase (PAH); diet; mutation; phenotype-genotype correlation; phenylketonuria (PKU); therapy.
Conflict of interest statement
Figures
Similar articles
-
Genetic etiology and clinical challenges of phenylketonuria.Hum Genomics. 2022 Jul 19;16(1):22. doi: 10.1186/s40246-022-00398-9. Hum Genomics. 2022. PMID: 35854334 Free PMC article. Review.
-
Long-term enzymatic and phenotypic correction in the phenylketonuria mouse model by adeno-associated virus vector-mediated gene transfer.Pediatr Res. 2004 Aug;56(2):278-84. doi: 10.1203/01.PDR.0000132837.29067.0E. Epub 2004 Jun 4. Pediatr Res. 2004. PMID: 15181195
-
Alternative therapies to address the unmet medical needs of patients with phenylketonuria.Expert Opin Pharmacother. 2015 Apr;16(6):791-800. doi: 10.1517/14656566.2015.1013030. Epub 2015 Feb 7. Expert Opin Pharmacother. 2015. PMID: 25660215
-
Phenylalanine hydroxylase genotype-phenotype associations in the United States: A single center study.Mol Genet Metab. 2019 Dec;128(4):415-421. doi: 10.1016/j.ymgme.2019.09.004. Epub 2019 Sep 14. Mol Genet Metab. 2019. PMID: 31623983
-
A structural hypothesis for BH4 responsiveness in patients with mild forms of hyperphenylalaninaemia and phenylketonuria.J Inherit Metab Dis. 2001 Apr;24(2):213-30. doi: 10.1023/a:1010371002631. J Inherit Metab Dis. 2001. PMID: 11405341 Review.
Cited by
-
Alchemical Design of Pharmacological Chaperones with Higher Affinity for Phenylalanine Hydroxylase.Int J Mol Sci. 2022 Apr 19;23(9):4502. doi: 10.3390/ijms23094502. Int J Mol Sci. 2022. PMID: 35562892 Free PMC article.
-
Phenylketonuria: a review of current and future treatments.Transl Pediatr. 2015 Oct;4(4):304-17. doi: 10.3978/j.issn.2224-4336.2015.10.07. Transl Pediatr. 2015. PMID: 26835392 Free PMC article. Review.
-
Expert opinion of an Italian working group on the assessment of cognitive, psychological, and neurological outcomes in pediatric, adolescent, and adult patients with phenylketonuria.Orphanet J Rare Dis. 2022 Dec 21;17(1):443. doi: 10.1186/s13023-022-02488-2. Orphanet J Rare Dis. 2022. PMID: 36544165 Free PMC article.
-
Inborn errors of amino acid metabolism - from underlying pathophysiology to therapeutic advances.Dis Model Mech. 2023 Nov 1;16(11):dmm050233. doi: 10.1242/dmm.050233. Epub 2023 Nov 23. Dis Model Mech. 2023. PMID: 37994477 Free PMC article.
-
Engineering Organoids for in vitro Modeling of Phenylketonuria.Front Mol Neurosci. 2022 Jan 10;14:787242. doi: 10.3389/fnmol.2021.787242. eCollection 2021. Front Mol Neurosci. 2022. PMID: 35082602 Free PMC article. Review.
References
-
- Scriver CR, Eisensmith RC, Woo SL, et al. The hyperphenylalaninemias of man and mouse. Annu Rev Genet 1994;28:141-65. - PubMed
-
- Penrose LS. Inheritance of phenylpyruvic amentia (Phenylketonuria). Lancet 1935;2:192-4.
-
- Guthrie R, Susi A. A simple phenylalanine method for detecting phenylketonuria in large populations of newborn infants. Pediatrics 1963;32:338-43. - PubMed
-
- Blau N, Van Spronsen FJ, Levy HL. Phenylketonuria. Lancet 2010;376:1417-27. - PubMed
-
- Thöny B, Blau N. Mutations in the BH4-metabolizing genes GTP cyclohydrolase I, 6-pyruvoyl-tetrahydropterin synthase, sepiapterin reductase, carbinolamine-4a-dehydratase, and dihydropteridine reductase. Hum Mutat 2006;27:870-8. - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources