

Retinal dystrophies, genomic applications in diagnosis and prospects for therapy
- PMID: 26835369
- PMCID: PMC4729094
- DOI: 10.3978/j.issn.2224-4336.2015.04.03
Retinal dystrophies, genomic applications in diagnosis and prospects for therapy
Abstract
Retinal dystrophies (RDs) are degenerative diseases of the retina which have marked clinical and genetic heterogeneity. Common presentations among these disorders include night or colour blindness, tunnel vision and subsequent progression to complete blindness. The known causative disease genes have a variety of developmental and functional roles with mutations in more than 120 genes shown to be responsible for the phenotypes. In addition, mutations within the same gene have been shown to cause different disease phenotypes, even amongst affected individuals within the same family highlighting further levels of complexity. The known disease genes encode proteins involved in retinal cellular structures, phototransduction, the visual cycle, and photoreceptor structure or gene regulation. This review aims to demonstrate the high degree of genetic complexity in both the causative disease genes and their associated phenotypes, highlighting the more common clinical manifestation of retinitis pigmentosa (RP). The review also provides insight to recent advances in genomic molecular diagnosis and gene and cell-based therapies for the RDs.
Keywords: CRISPR/Cas9; Retinal dystrophy (RD); massively parallel sequencing (MPS); retinitis pigmentosa (RP).
Conflict of interest statement
Figures
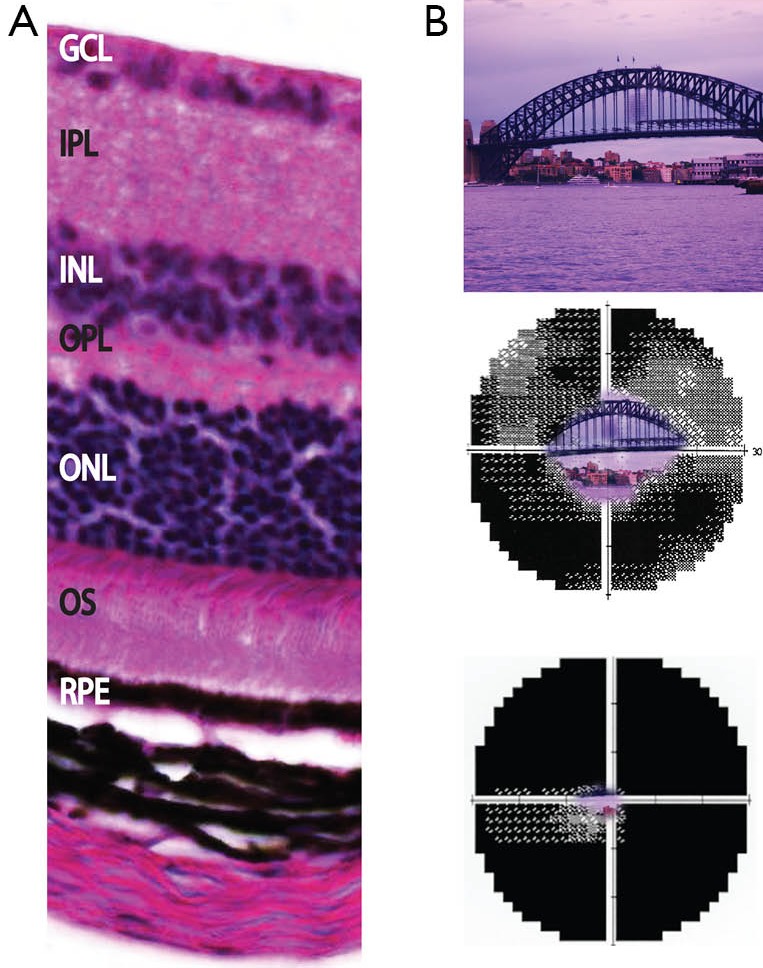
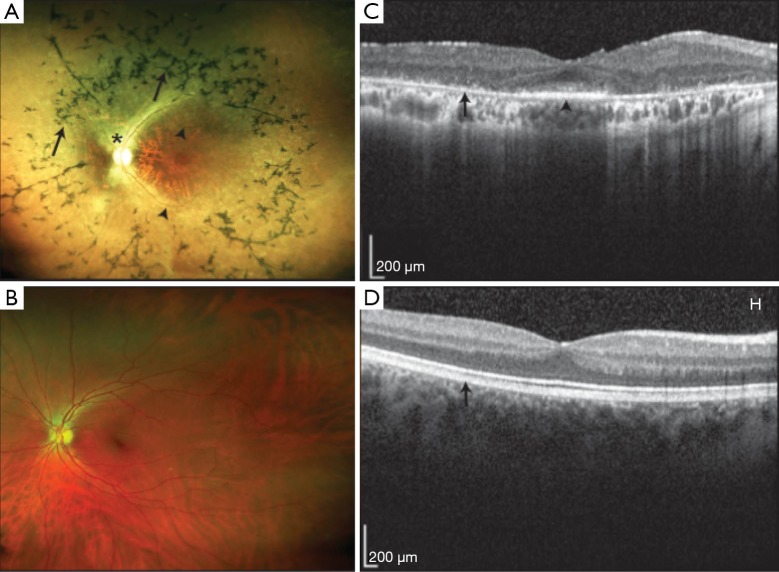
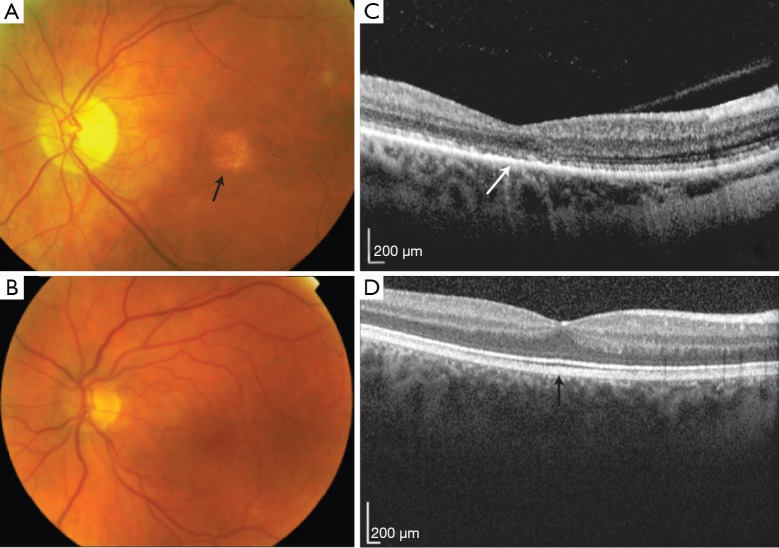
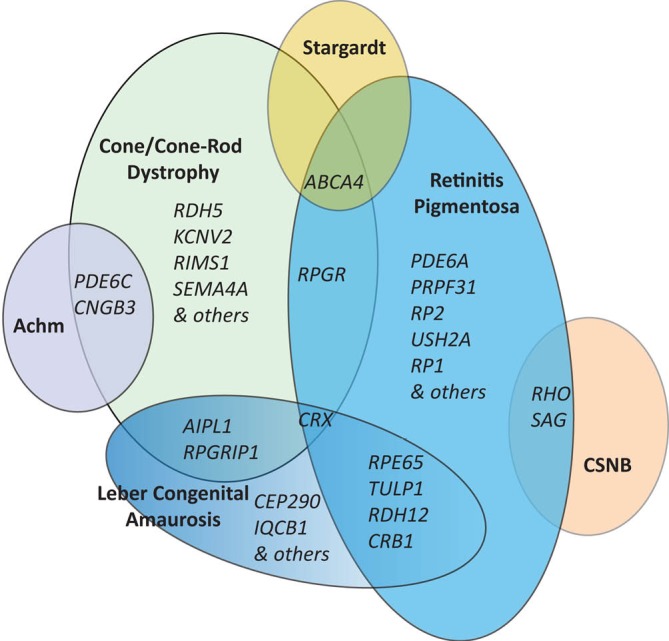
References
-
- Haim M. The epidemiology of retinitis pigmentosa in Denmark. Acta Ophthalmol Scand Suppl 2002;(233):1-34. - PubMed
-
- Martínez-Mir A, Paloma E, Allikmets R, et al. Retinitis pigmentosa caused by a homozygous mutation in the Stargardt disease gene ABCR. Nat Genet 1998;18:11-2. - PubMed
-
- Meindl A, Dry K, Herrmann K, Manson F, et al. A gene (RPGR) with homology to the RCC1 guanine nucleotide exchange factor is mutated in X-linked retinitis pigmentosa (RP3). Nat Genet 1996;13:35-42. - PubMed
-
- Daiger SP, Rossiter BJ, Greenberg J, et al. RetNet - Retinal Information Network 1998. Data services and software for identifying genes and mutations causing retinal degeneration]. Available online: http://www.sph.uth.tmc.edu/RetNet/ [updated Dec 08, 2014; cited Jan 23, 2015].
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous