

Investigation and management of the hepatic glycogen storage diseases
- PMID: 26835382
- PMCID: PMC4729058
- DOI: 10.3978/j.issn.2224-4336.2015.04.07
Investigation and management of the hepatic glycogen storage diseases
Abstract
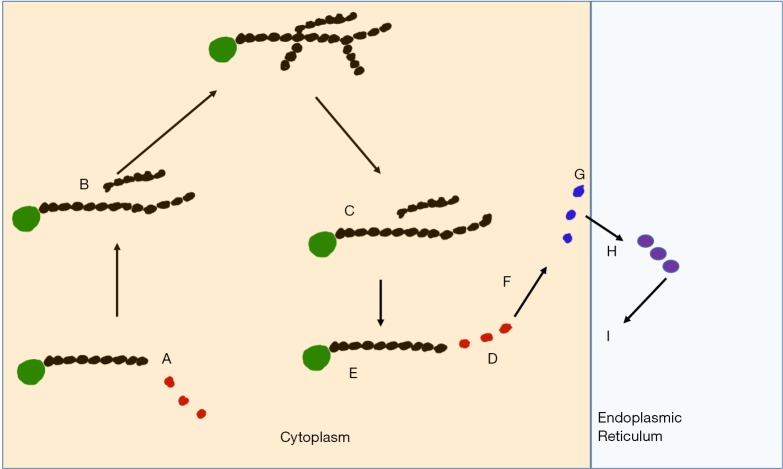
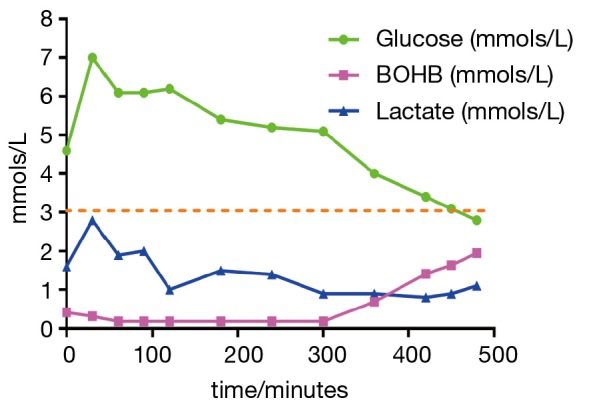
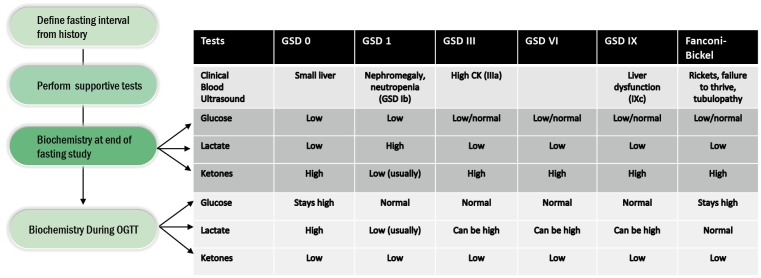
The glycogen storage diseases (GSD) comprise a group of disorders that involve the disruption of metabolism of glycogen. Glycogen is stored in various organs including skeletal muscle, the kidneys and liver. The liver stores glycogen to supply the rest of the body with glucose when required. Therefore, disruption of this process can lead to hypoglycaemia. If glycogen is not broken down effectively, this can lead to hepatomegaly. Glycogen synthase deficiency leads to impaired glycogen synthesis and consequently the liver is small. Glycogen brancher deficiency can lead to abnormal glycogen being stored in the liver leading to a quite different disorder of progressive liver dysfunction. Understanding the physiology of GSD I, III, VI and IX guides dietary treatments and the provision of appropriate amounts and types of carbohydrates. There has been recent re-emergence in the literature of the use of ketones in therapy, either in the form of the salt D,L-3-hydroxybutyrate or medium chain triglyceride (MCT). High protein diets have also been advocated. Alternative waxy maize based starches seem to show promising early data of efficacy. There are many complications of each of these disorders and they need to be prospectively surveyed and managed. Liver and kidney transplantation is still indicated in severe refractory disease.
Keywords: Glycogen storage disease (GSD); Glycosade; hepatomegaly; hypoglycaemia; ketones; lactate; medium chain triglyceride (MCT); next generation sequencing; waxy maize starch.
Conflict of interest statement
Figures
Similar articles
-
Nutrition therapy for hepatic glycogen storage diseases.J Am Diet Assoc. 1993 Dec;93(12):1423-30. doi: 10.1016/0002-8223(93)92246-t. J Am Diet Assoc. 1993. PMID: 8245377 Review.
-
Short and long-term acceptability and efficacy of extended-release cornstarch in the hepatic glycogen storage diseases: results from the Glyde study.Orphanet J Rare Dis. 2024 Jul 9;19(1):258. doi: 10.1186/s13023-024-03274-y. Orphanet J Rare Dis. 2024. PMID: 38982397 Free PMC article. Clinical Trial.
-
Hepatic glycogen storage disorders: what have we learned in recent years?Curr Opin Clin Nutr Metab Care. 2015 Jul;18(4):415-21. doi: 10.1097/MCO.0000000000000181. Curr Opin Clin Nutr Metab Care. 2015. PMID: 26001652 Review.
-
Glycogen storage diseases: new perspectives.World J Gastroenterol. 2007 May 14;13(18):2541-53. doi: 10.3748/wjg.v13.i18.2541. World J Gastroenterol. 2007. PMID: 17552001 Free PMC article. Review.
-
Glycogen metabolism and glycogen storage disorders.Ann Transl Med. 2018 Dec;6(24):474. doi: 10.21037/atm.2018.10.59. Ann Transl Med. 2018. PMID: 30740405 Free PMC article. Review.
Cited by
-
Hypoglycaemia Metabolic Gene Panel Testing.Front Endocrinol (Lausanne). 2022 Mar 29;13:826167. doi: 10.3389/fendo.2022.826167. eCollection 2022. Front Endocrinol (Lausanne). 2022. PMID: 35422763 Free PMC article. Review.
-
Platelet glycogenolysis is important for energy production and function.Platelets. 2023 Dec;34(1):2222184. doi: 10.1080/09537104.2023.2222184. Platelets. 2023. PMID: 37292023 Free PMC article.
-
A case study of a liver transplant-treated patient with glycogen storage disease type Ia presenting with multiple inflammatory hepatic adenomas: an analysis of clinicopathologic and genetic data.BMC Med Genomics. 2024 May 6;17(1):124. doi: 10.1186/s12920-024-01888-6. BMC Med Genomics. 2024. PMID: 38711024 Free PMC article.
-
Development of minimally invasive 13C-glucose breath test to examine different exogenous carbohydrate sources in patients with glycogen storage disease type Ia.Mol Genet Metab Rep. 2022 May 11;31:100880. doi: 10.1016/j.ymgmr.2022.100880. eCollection 2022 Jun. Mol Genet Metab Rep. 2022. PMID: 35585965 Free PMC article.
-
Identification of Potential Therapeutic Targets Against Anthrax-Toxin-Induced Liver and Heart Damage.Toxins (Basel). 2025 Jan 24;17(2):54. doi: 10.3390/toxins17020054. Toxins (Basel). 2025. PMID: 39998071 Free PMC article.
References
-
- Lee PJ, Bhattacharya K. Glycogen Storage Diseases. Oxford: Oxford University Press, 2013. Available online: http://oxfordmedicine.com/view/10.1093/med/9780199204854.001.1/med-97801... - DOI
-
- Rake JP, Visser G, Labrune P, et al. Glycogen storage disease type I: diagnosis, management, clinical course and outcome. Results of the European Study on Glycogen Storage Disease Type I (ESGSD I). Eur J Pediatr 2002;161 Suppl 1:S20-34. - PubMed
-
- Schippers HM, Smit GP, Rake JP, et al. Characteristic growth pattern in male X-linked phosphorylase-b kinase deficiency (GSD IX). J Inherit Metab Dis 2003;26:43-7. - PubMed
-
- Kishnani PS, Austin SL, Arn P, et al. Glycogen storage disease type III diagnosis and management guidelines. Genet Med 2010;12:446-63. - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources