

Phenylketonuria: a review of current and future treatments
- PMID: 26835392
- PMCID: PMC4728993
- DOI: 10.3978/j.issn.2224-4336.2015.10.07
Phenylketonuria: a review of current and future treatments
Abstract
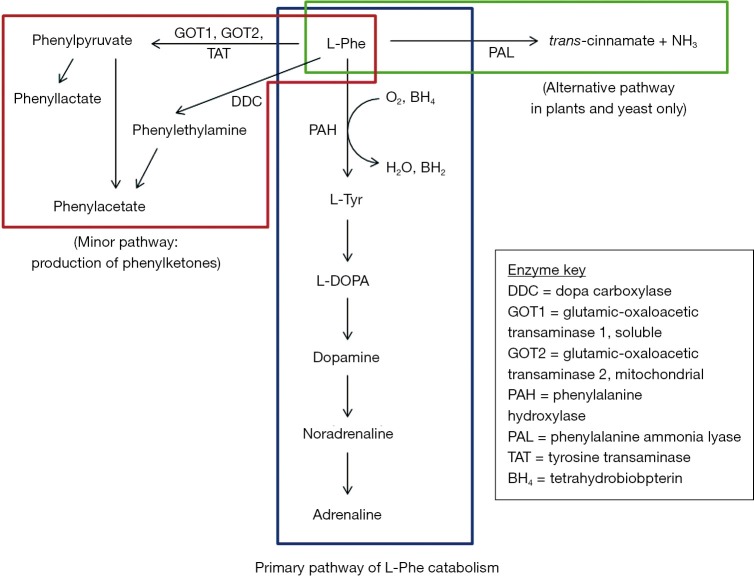
Phenylketonuria (PKU) is an autosomal recessive inborn error of metabolism caused by a deficiency in the hepatic enzyme phenylalanine hydroxylase (PAH). If left untreated, the main clinical feature is intellectual disability. Treatment, which includes a low Phe diet supplemented with amino acid formulas, commences soon after diagnosis within the first weeks of life. Although dietary treatment has been successful in preventing intellectual disability in early treated PKU patients, there are major issues with dietary compliance due to palatability of the diet. Other potential issues associated with dietary therapy include nutritional deficiencies especially vitamin D and B12. Suboptimal outcomes in cognitive and executive functioning have been reported in patients who adhere poorly to dietary therapy. There have been continuous attempts at improving the quality of medical foods including their palatability. Advances in dietary therapy such as the use of large neutral amino acids (LNAA) and glycomacropeptides (GMP; found within the whey fraction of bovine milk) have been explored. Gene therapy and enzyme replacement or substitution therapy have yielded more promising data in the recent years. In this review the current and possible future treatments for PKU are discussed.
Keywords: Phenylketonuria (PKU); dietary therapy; glycomacropeptides (GMP); large neutral amino acids (LNAA); phenylalanine ammonia lyase; phenylalanine hydroxylase (PAH); probiotic; tetrahydrobiopterin.
Conflict of interest statement
Figures
References
-
- Scriver CR, Kaufman S. Hyperphenylalaninemia: phenylalanine hydroxylase deficiency. In: Scriver CR, Beaudet AL, Sly SW, et al. editors. The Metabolic and Molecular Bases of Inherited Disease. 8 ed. New York, NY: McGraw-Hill, 2001:1667-724.
-
- Blau N, van Spronsen FJ, Levy HL. Phenylketonuria. Lancet 2010;376:1417-27. - PubMed
-
- Matalon R, Michals K. Phenylketonuria: screening, treatment and maternal PKU. Clin Biochem 1991;24:337-42. - PubMed
-
- Loeber JG. Neonatal screening in Europe; the situation in 2004. J Inherit Metab Dis 2007;30:430-8. - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources