

Fragmentation trees reloaded
- PMID: 26839597
- PMCID: PMC4736045
- DOI: 10.1186/s13321-016-0116-8
Fragmentation trees reloaded
Abstract
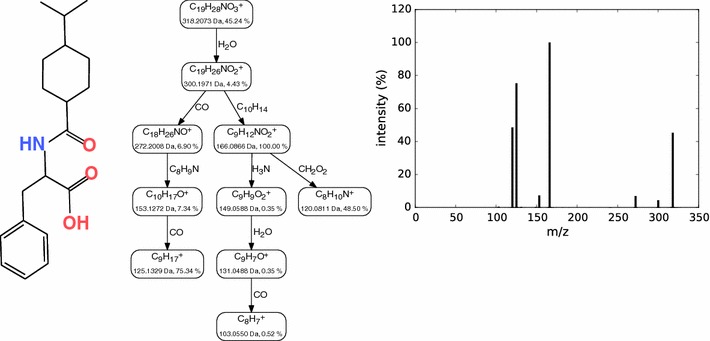
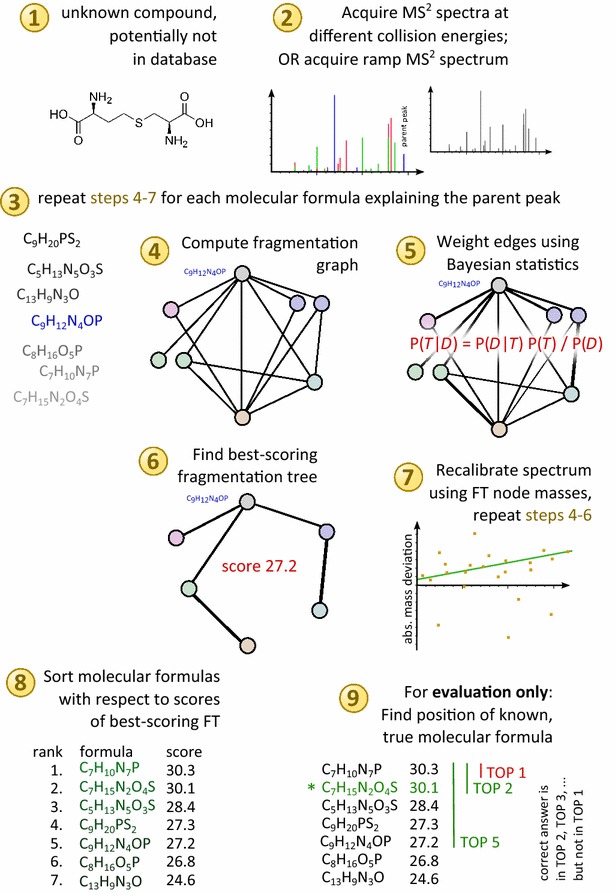
Background: Untargeted metabolomics commonly uses liquid chromatography mass spectrometry to measure abundances of metabolites; subsequent tandem mass spectrometry is used to derive information about individual compounds. One of the bottlenecks in this experimental setup is the interpretation of fragmentation spectra to accurately and efficiently identify compounds. Fragmentation trees have become a powerful tool for the interpretation of tandem mass spectrometry data of small molecules. These trees are determined from the data using combinatorial optimization, and aim at explaining the experimental data via fragmentation cascades. Fragmentation tree computation does not require spectral or structural databases. To obtain biochemically meaningful trees, one needs an elaborate optimization function (scoring).
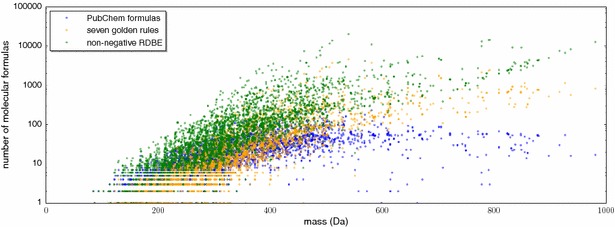
Results: We present a new scoring for computing fragmentation trees, transforming the combinatorial optimization into a Maximum A Posteriori estimator. We demonstrate the superiority of the new scoring for two tasks: both for the de novo identification of molecular formulas of unknown compounds, and for searching a database for structurally similar compounds, our method SIRIUS 3, performs significantly better than the previous version of our method, as well as other methods for this task.
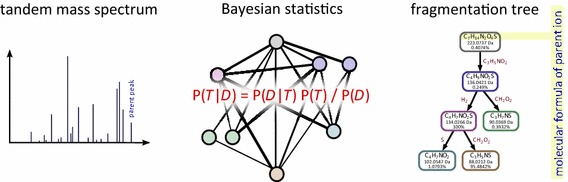
Conclusion: SIRIUS 3 can be a part of an untargeted metabolomics workflow, allowing researchers to investigate unknowns using automated computational methods.Graphical abstractWe present a new scoring for computing fragmentation trees from tandem mass spectrometry data based on Bayesian statistics. The best scoring fragmentation tree most likely explains the molecular formula of the measured parent ion.
Keywords: Computational methods; Fragmentation trees; Mass spectrometry; Metabolites; Natural products.
Figures
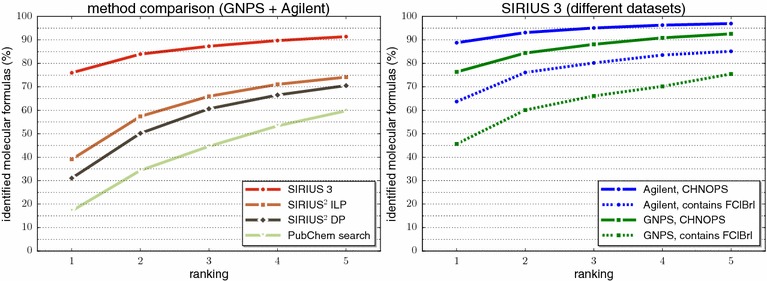
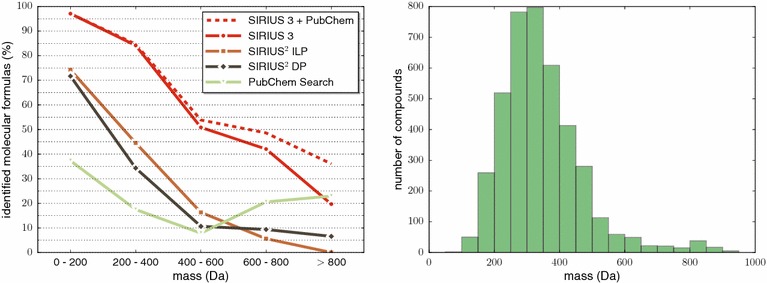
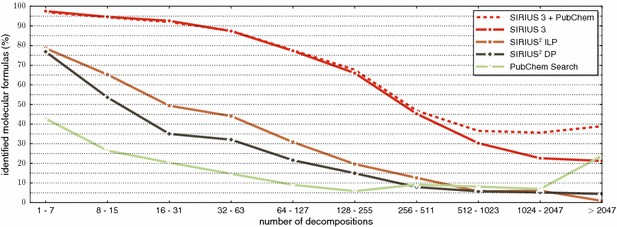
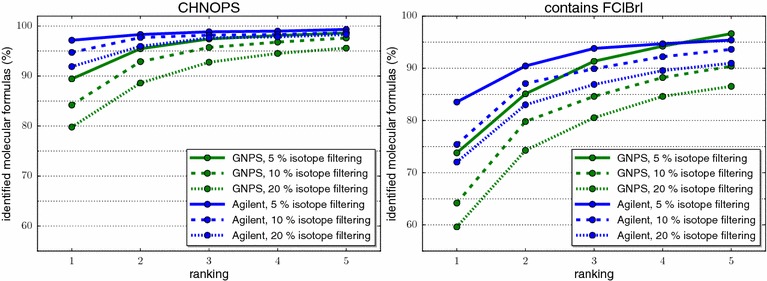
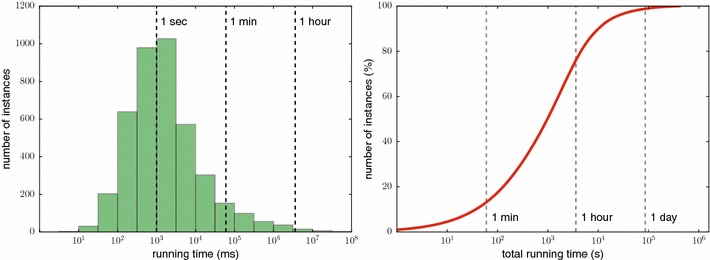
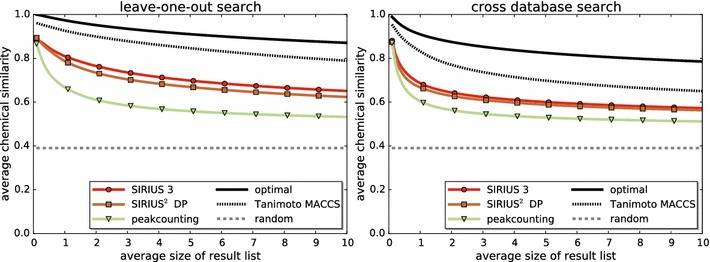
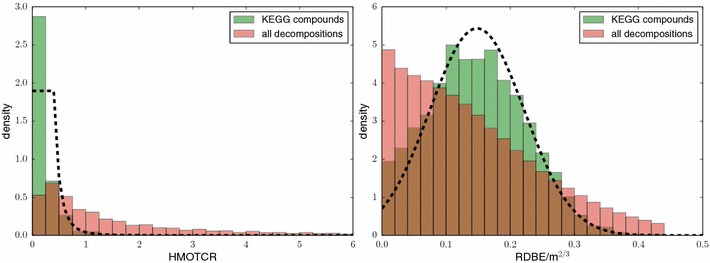
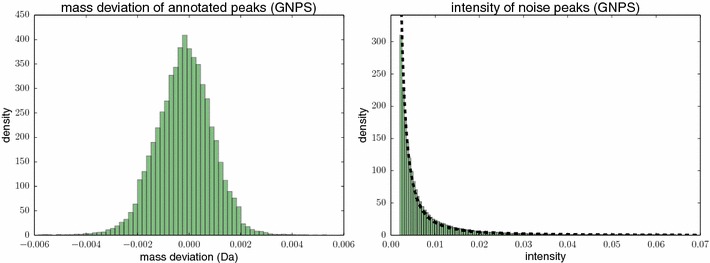
References
-
- Baker M. Metabolomics: from small molecules to big ideas. Nat Methods. 2011;8:117–121. doi: 10.1038/nmeth0211-117. - DOI
LinkOut - more resources
Full Text Sources
Other Literature Sources
