

Therapeutic Benefits of Induced Pluripotent Stem Cells in Monocrotaline-Induced Pulmonary Arterial Hypertension
- PMID: 26840075
- PMCID: PMC4740504
- DOI: 10.1371/journal.pone.0142476
Therapeutic Benefits of Induced Pluripotent Stem Cells in Monocrotaline-Induced Pulmonary Arterial Hypertension
Abstract
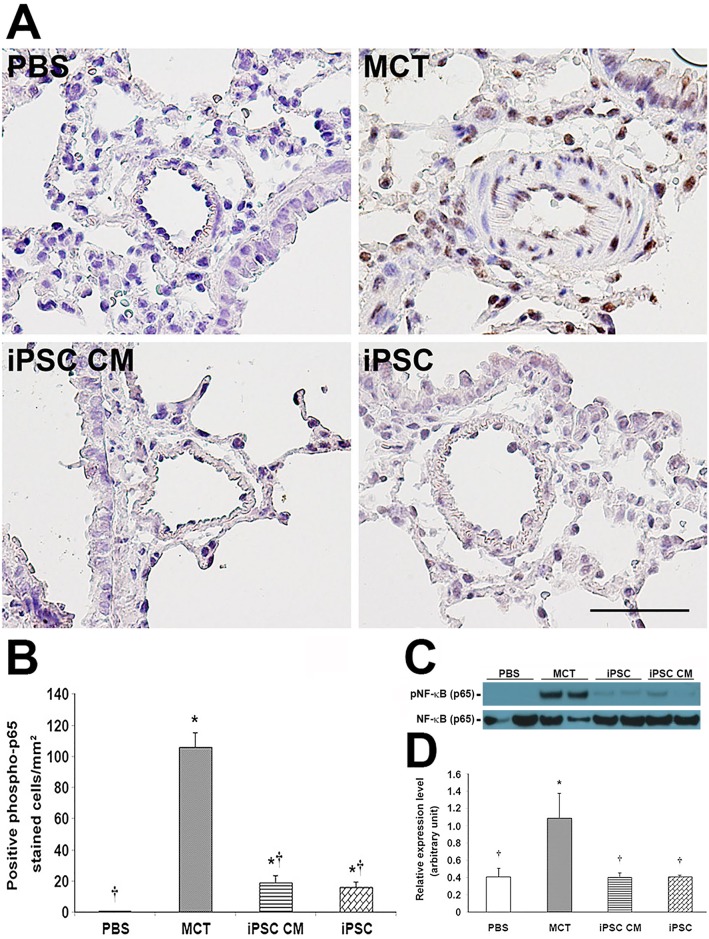
Pulmonary arterial hypertension (PAH) is characterized by progressive increases in vascular resistance and the remodeling of pulmonary arteries. The accumulation of inflammatory cells in the lung and elevated levels of inflammatory cytokines in the bloodstream suggest that inflammation may play a role in PAH. In this study, the benefits of induced pluripotent stem cells (iPSCs) and iPSC-conditioned medium (iPSC CM) were explored in monocrotaline (MCT)-induced PAH rats. We demonstrated that both iPSCs and iPSC CM significantly reduced the right ventricular systolic pressure and ameliorated the hypertrophy of the right ventricle in MCT-induced PAH rats in models of both disease prevention and disease reversal. In the prevention of MCT-induced PAH, iPSC-based therapy led to the decreased accumulation of inflammatory cells and down-regulated the expression of the IL-1β, IL-6, IL-12α, IL-12β, IL-23 and IFNγ genes in lung specimens, which implied that iPSC-based therapy may be involved in the regulation of inflammation. NF-κB signaling is essential to the inflammatory cascade, which is activated via the phosphorylation of the NF-κB molecule. Using the chemical inhibitor specifically blocked the phosphorylation of NF-κB, and in vitro assays of cultured human M1 macrophages implied that the anti-inflammation effect of iPSC-based therapy may contribute to the disturbance of NF-κB activation. Here, we showed that iPSC-based therapy could restore the hemodynamic function of right ventricle with benefits for preventing the ongoing inflammation in the lungs of MCT-induced PAH rats by regulating NF-κB phosphorylation.
Conflict of interest statement
Figures
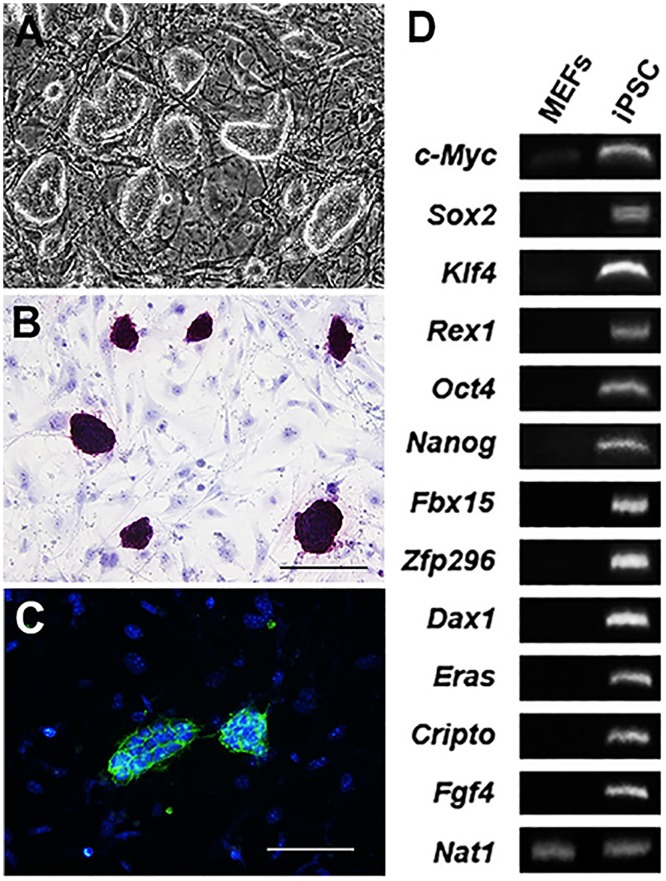
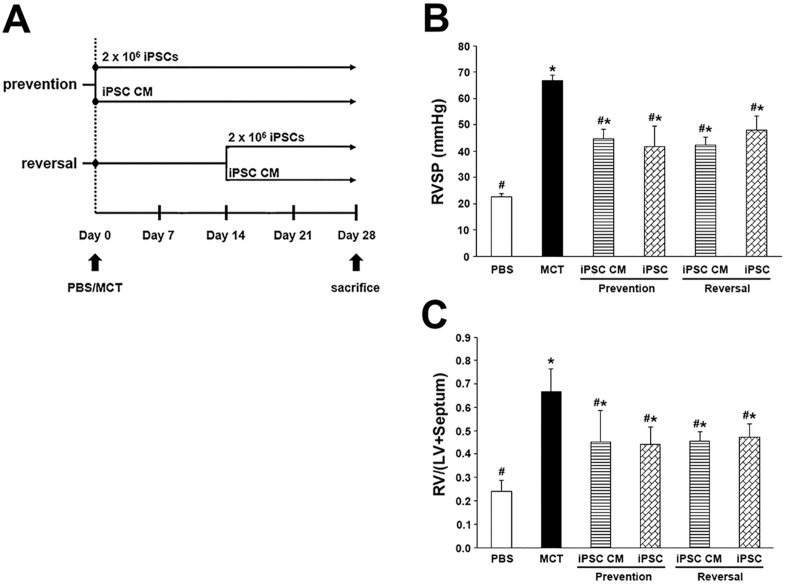
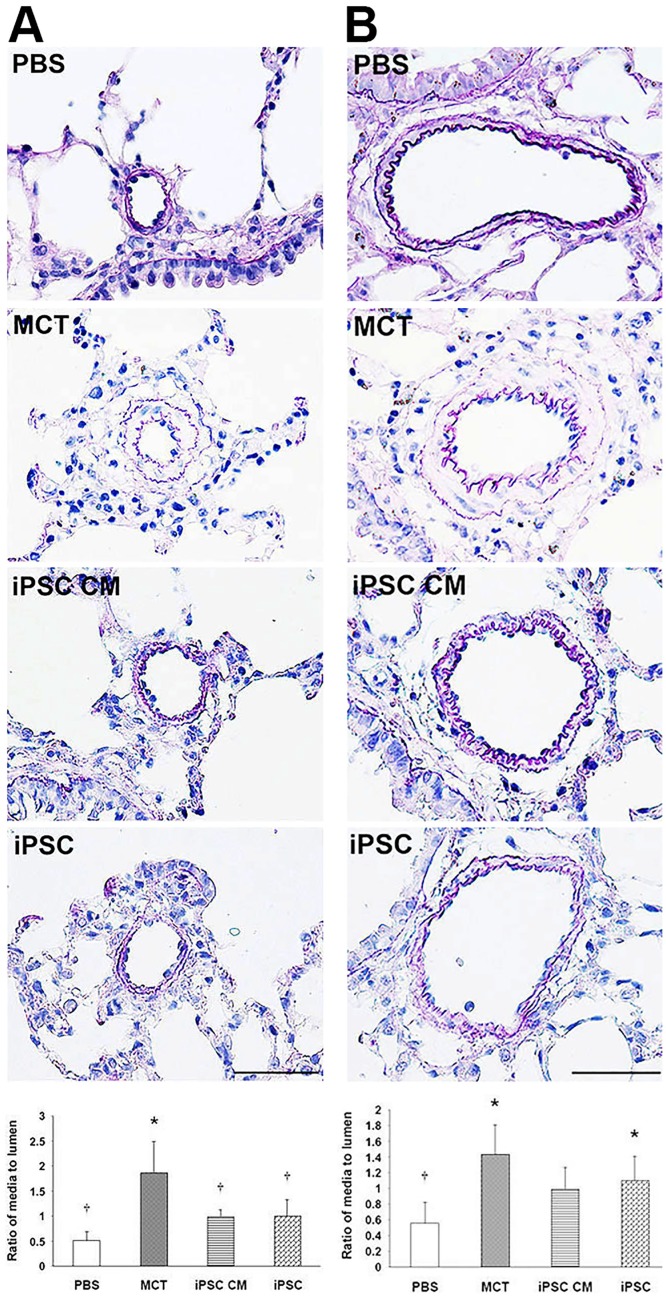

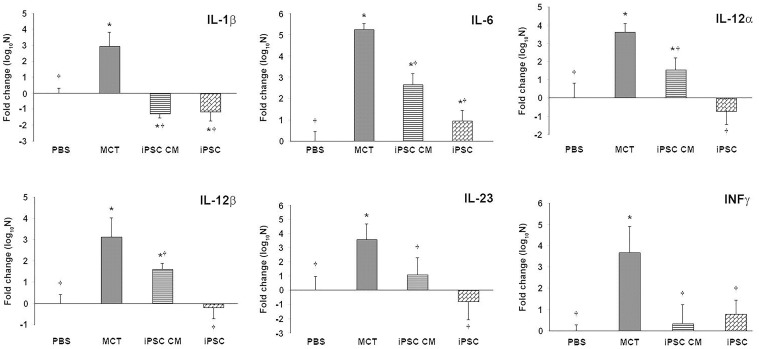

References
-
- Humbert M, Morrell NW, Archer SL, Stenmark KR, MacLean MR, Lang IM, et al. Cellular and molecular pathobiology of pulmonary arterial hypertension. J Am Coll Cardiol. 2004;43 (12 Suppl S): 13S–24S. - PubMed
-
- Budhiraja R, Tuder RM, Hassoun PM. Endothelial dysfunction in pulmonary hypertension. Circulation. 2004;109(2): 159–65. - PubMed