

The Distribution of Prion Protein Allotypes Differs Between Sporadic and Iatrogenic Creutzfeldt-Jakob Disease Patients
- PMID: 26840342
- PMCID: PMC4740439
- DOI: 10.1371/journal.ppat.1005416
The Distribution of Prion Protein Allotypes Differs Between Sporadic and Iatrogenic Creutzfeldt-Jakob Disease Patients
Erratum in
-
Correction: The Distribution of Prion Protein Allotypes Differs Between Sporadic and Iatrogenic Creutzfeldt-Jakob Disease Patients.PLoS Pathog. 2016 Mar 8;12(3):e1005496. doi: 10.1371/journal.ppat.1005496. eCollection 2016 Mar. PLoS Pathog. 2016. PMID: 26954665 Free PMC article. No abstract available.
Abstract
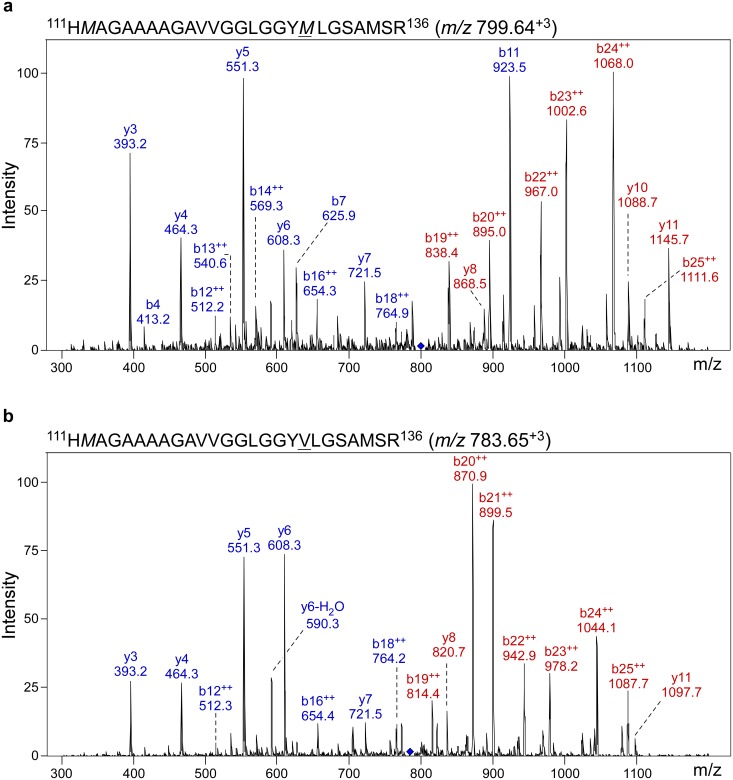
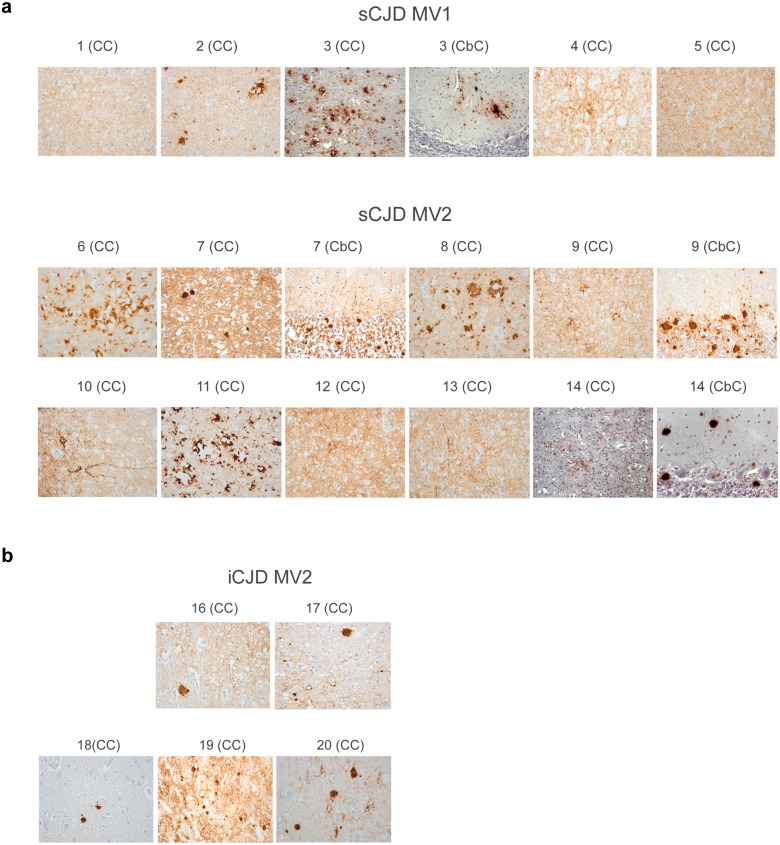
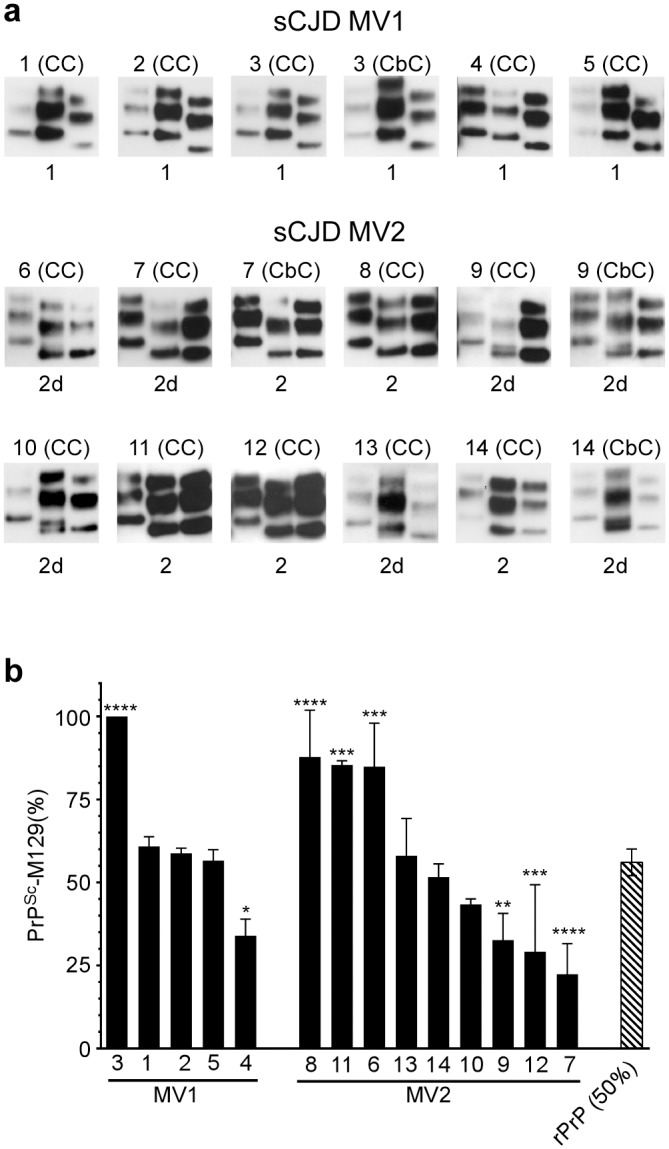
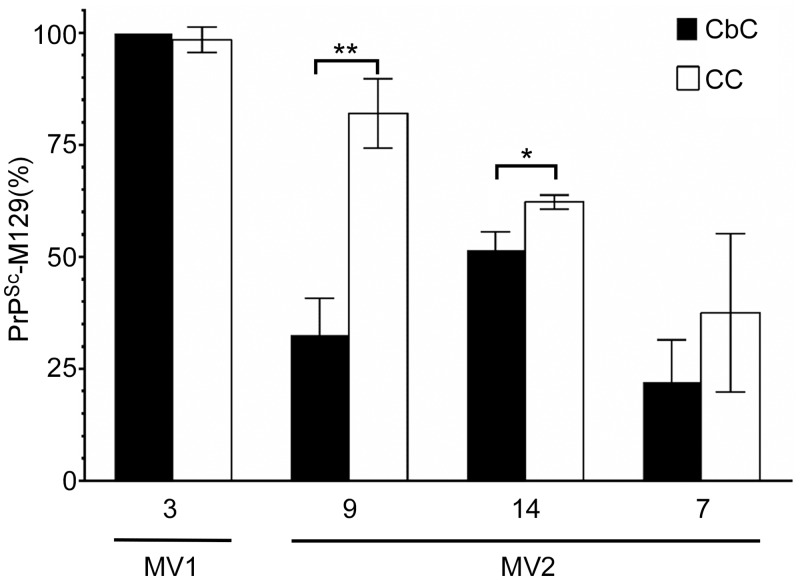
Sporadic Creutzfeldt-Jakob disease (sCJD) is the most prevalent of the human prion diseases, which are fatal and transmissible neurodegenerative diseases caused by the infectious prion protein (PrP(Sc)). The origin of sCJD is unknown, although the initiating event is thought to be the stochastic misfolding of endogenous prion protein (PrP(C)) into infectious PrP(Sc). By contrast, human growth hormone-associated cases of iatrogenic CJD (iCJD) in the United Kingdom (UK) are associated with exposure to an exogenous source of PrP(Sc). In both forms of CJD, heterozygosity at residue 129 for methionine (M) or valine (V) in the prion protein gene may affect disease phenotype, onset and progression. However, the relative contribution of each PrP(C) allotype to PrP(Sc) in heterozygous cases of CJD is unknown. Using mass spectrometry, we determined that the relative abundance of PrP(Sc) with M or V at residue 129 in brain specimens from MV cases of sCJD was highly variable. This result is consistent with PrP(C) containing an M or V at residue 129 having a similar propensity to misfold into PrP(Sc) thus causing sCJD. By contrast, PrP(Sc) with V at residue 129 predominated in the majority of the UK human growth hormone associated iCJD cases, consistent with exposure to infectious PrP(Sc) containing V at residue 129. In both types of CJD, the PrP(Sc) allotype ratio had no correlation with CJD type, age at clinical onset, or disease duration. Therefore, factors other than PrP(Sc) allotype abundance must influence the clinical progression and phenotype of heterozygous cases of CJD.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
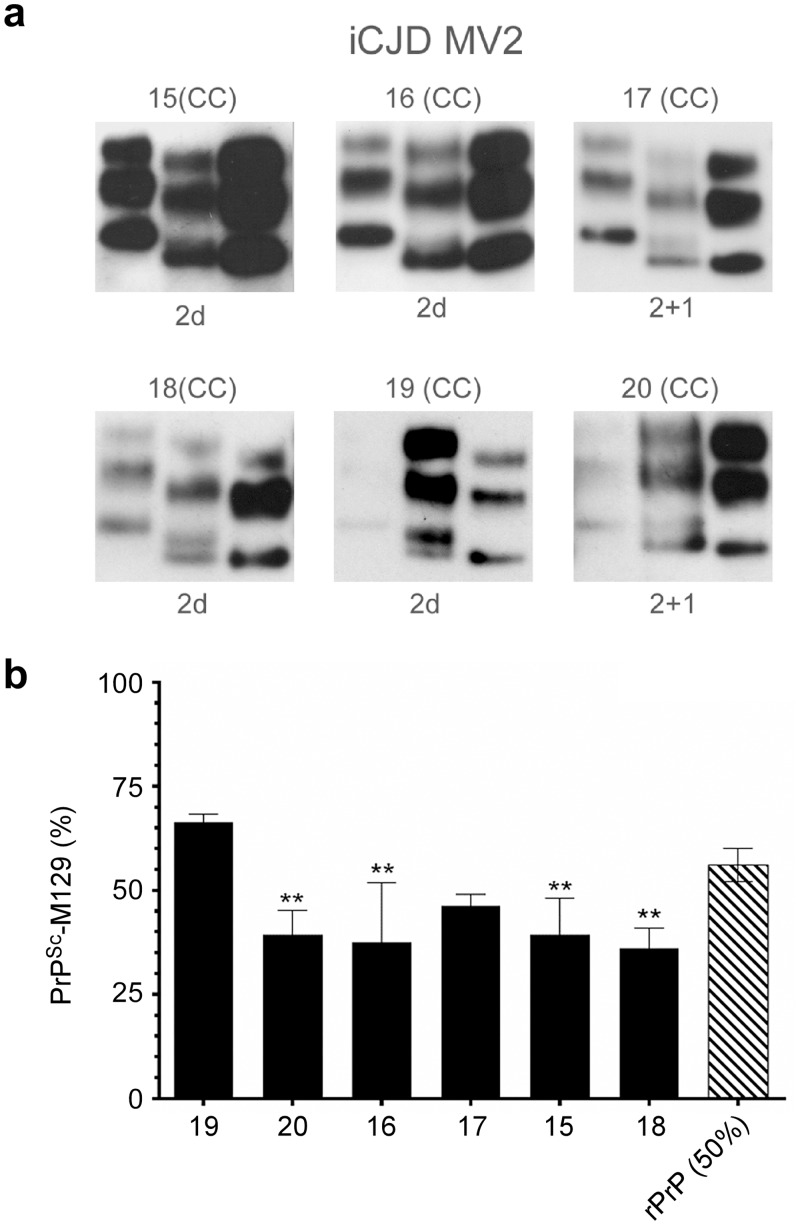
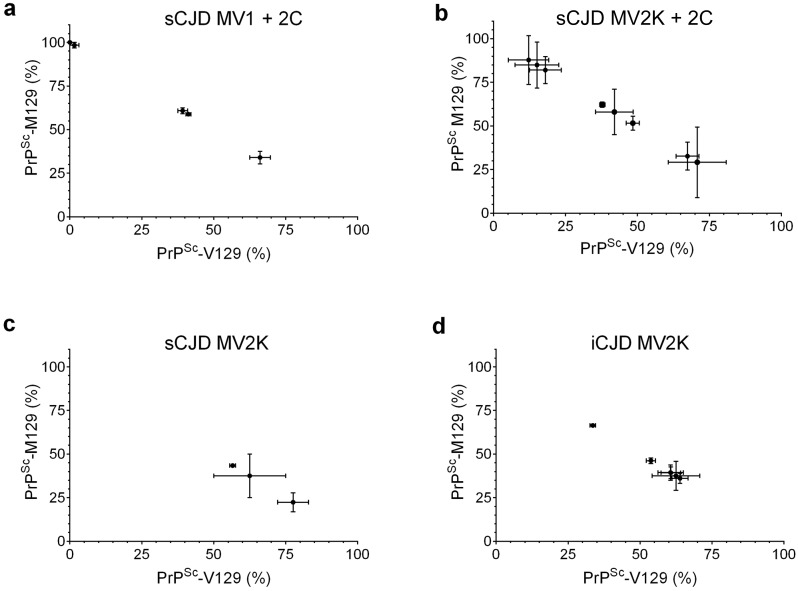
References
-
- Parchi P, Saverioni D. Molecular pathology, classification, and diagnosis of sporadic human prion disease variants. Folia Neuropathol. 2012;50: 20–45. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
