

Real-time, portable genome sequencing for Ebola surveillance
- PMID: 26840485
- PMCID: PMC4817224
- DOI: 10.1038/nature16996
Real-time, portable genome sequencing for Ebola surveillance
Abstract
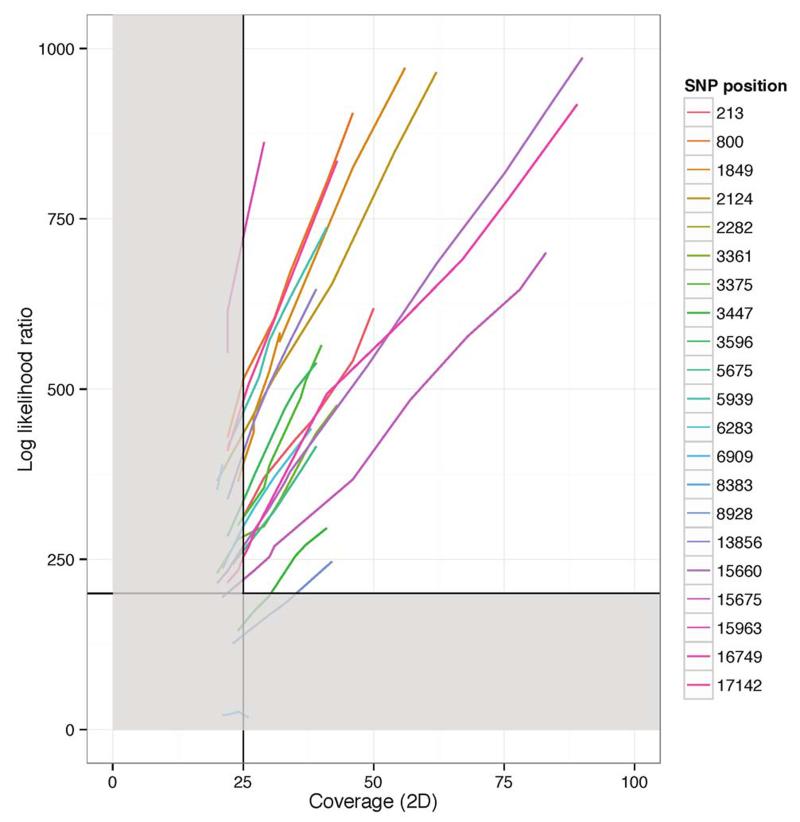
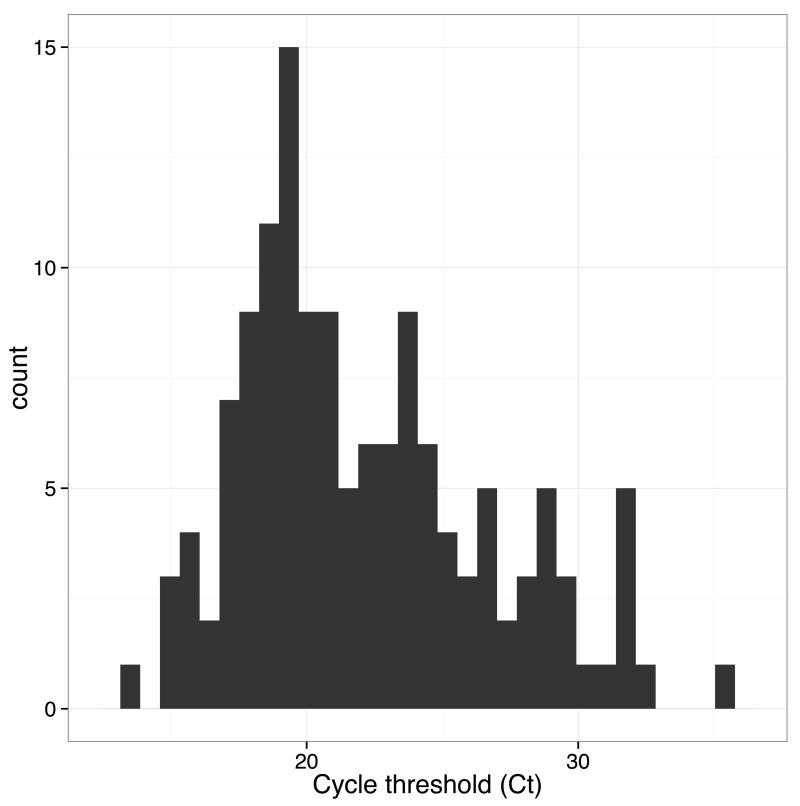
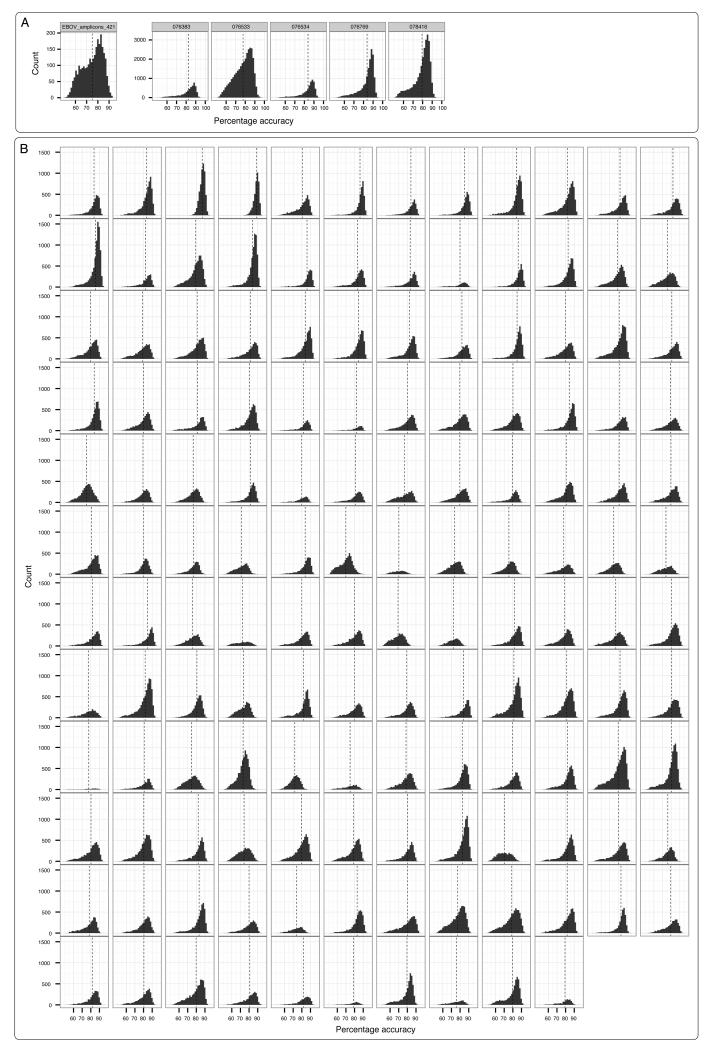
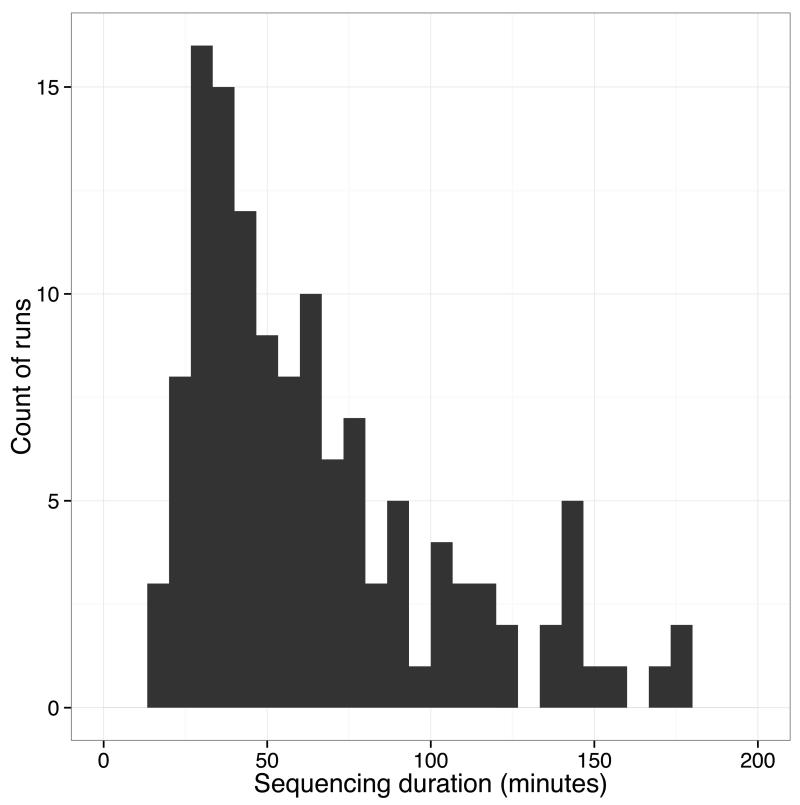
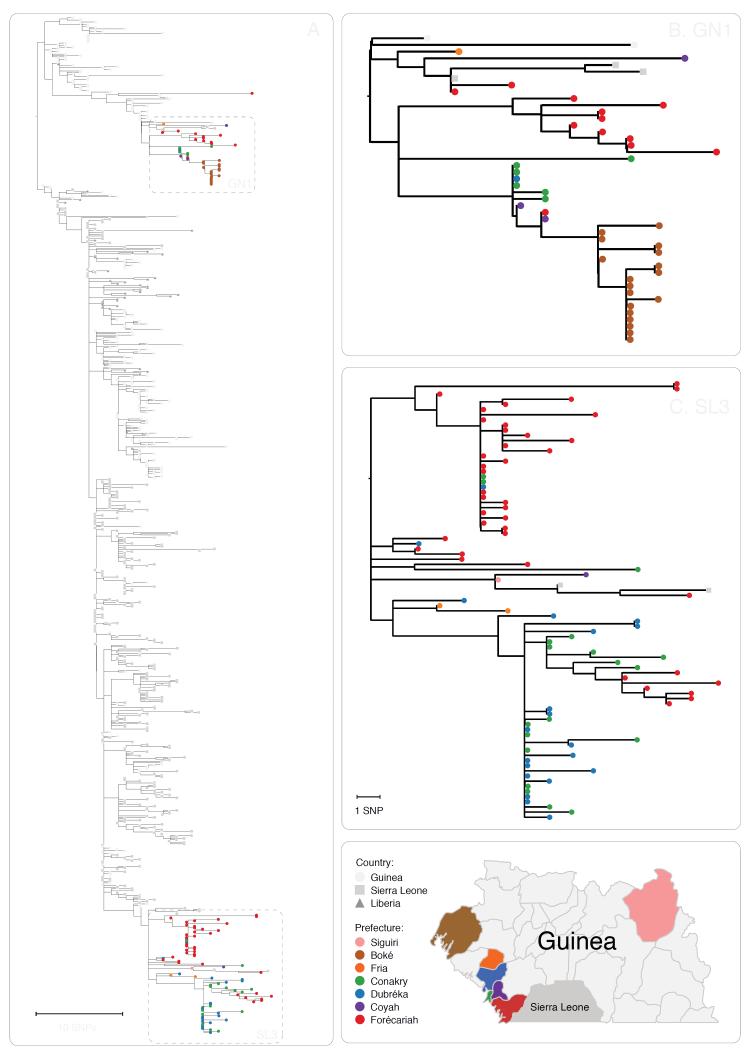
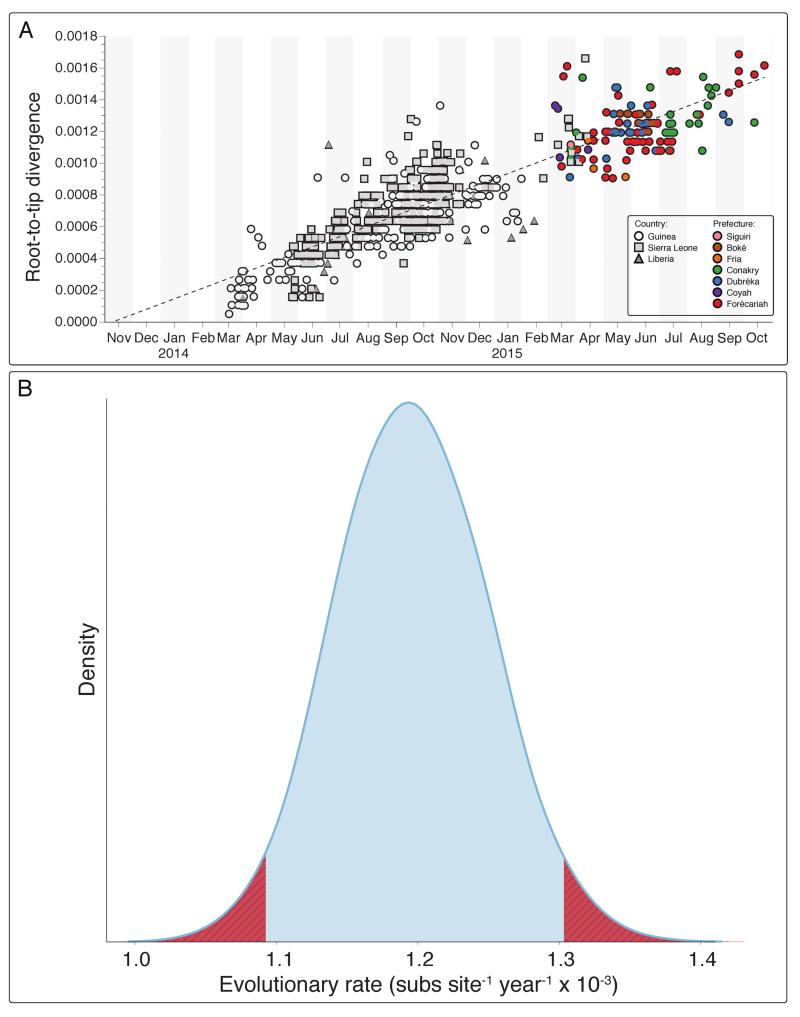
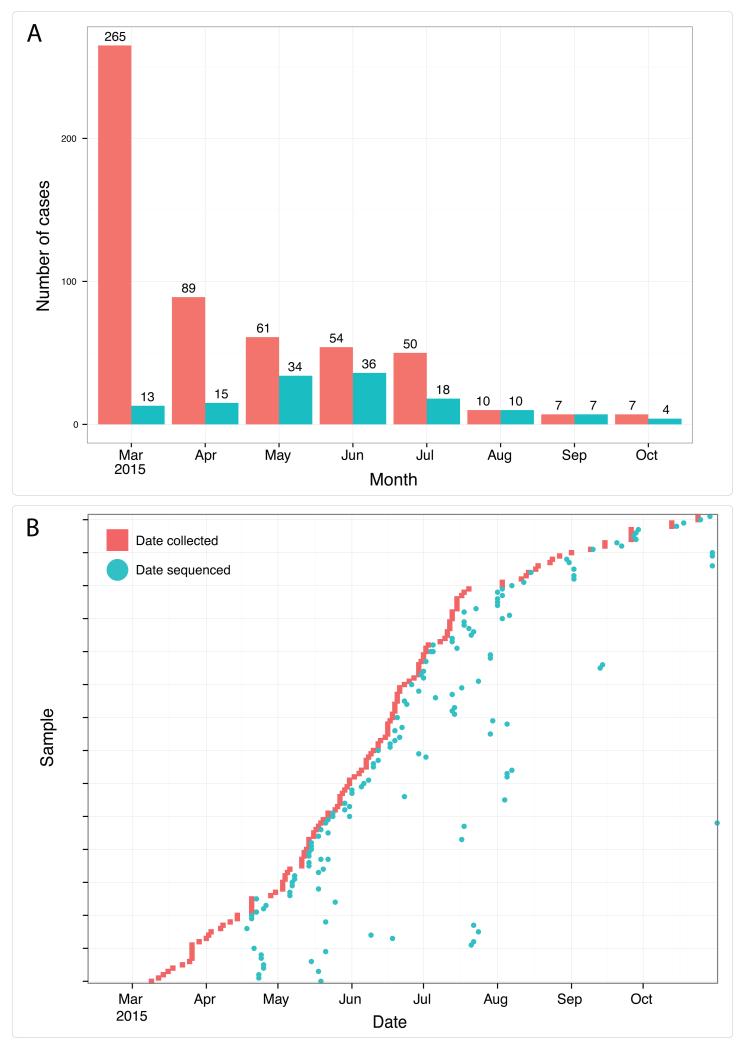
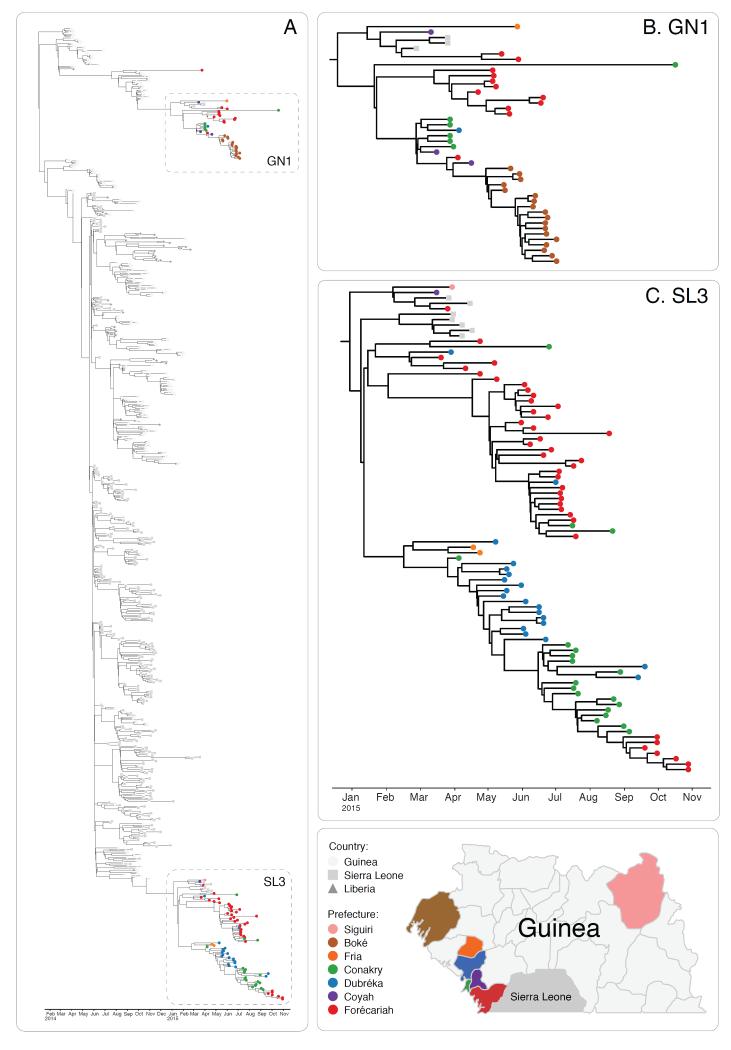
The Ebola virus disease epidemic in West Africa is the largest on record, responsible for over 28,599 cases and more than 11,299 deaths. Genome sequencing in viral outbreaks is desirable to characterize the infectious agent and determine its evolutionary rate. Genome sequencing also allows the identification of signatures of host adaptation, identification and monitoring of diagnostic targets, and characterization of responses to vaccines and treatments. The Ebola virus (EBOV) genome substitution rate in the Makona strain has been estimated at between 0.87 × 10(-3) and 1.42 × 10(-3) mutations per site per year. This is equivalent to 16-27 mutations in each genome, meaning that sequences diverge rapidly enough to identify distinct sub-lineages during a prolonged epidemic. Genome sequencing provides a high-resolution view of pathogen evolution and is increasingly sought after for outbreak surveillance. Sequence data may be used to guide control measures, but only if the results are generated quickly enough to inform interventions. Genomic surveillance during the epidemic has been sporadic owing to a lack of local sequencing capacity coupled with practical difficulties transporting samples to remote sequencing facilities. To address this problem, here we devise a genomic surveillance system that utilizes a novel nanopore DNA sequencing instrument. In April 2015 this system was transported in standard airline luggage to Guinea and used for real-time genomic surveillance of the ongoing epidemic. We present sequence data and analysis of 142 EBOV samples collected during the period March to October 2015. We were able to generate results less than 24 h after receiving an Ebola-positive sample, with the sequencing process taking as little as 15-60 min. We show that real-time genomic surveillance is possible in resource-limited settings and can be established rapidly to monitor outbreaks.
Figures
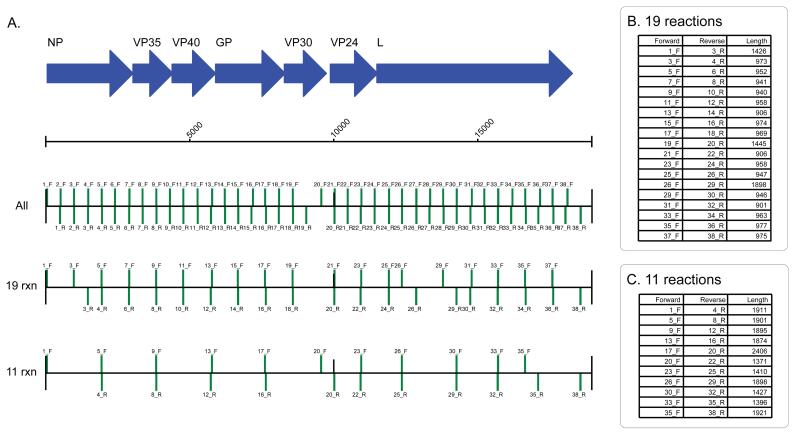
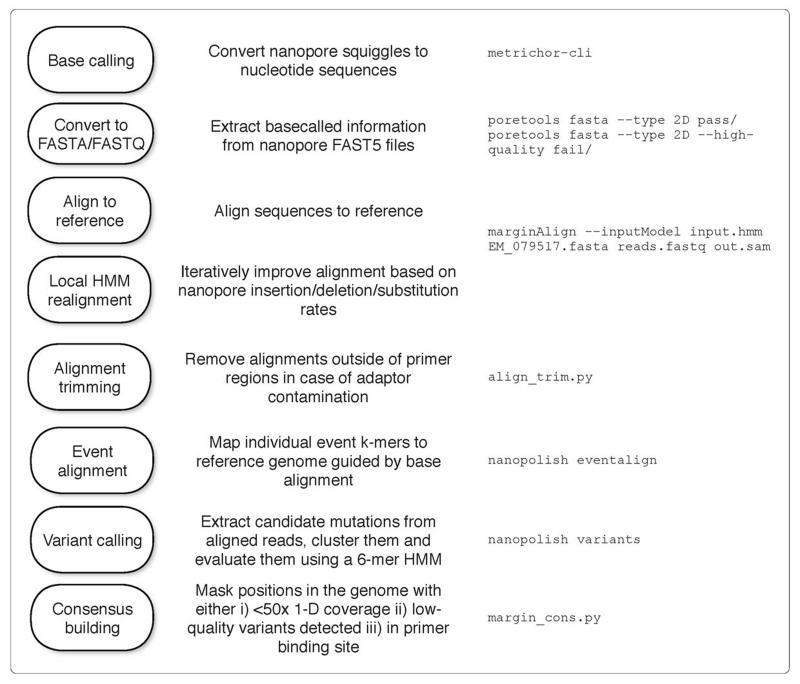
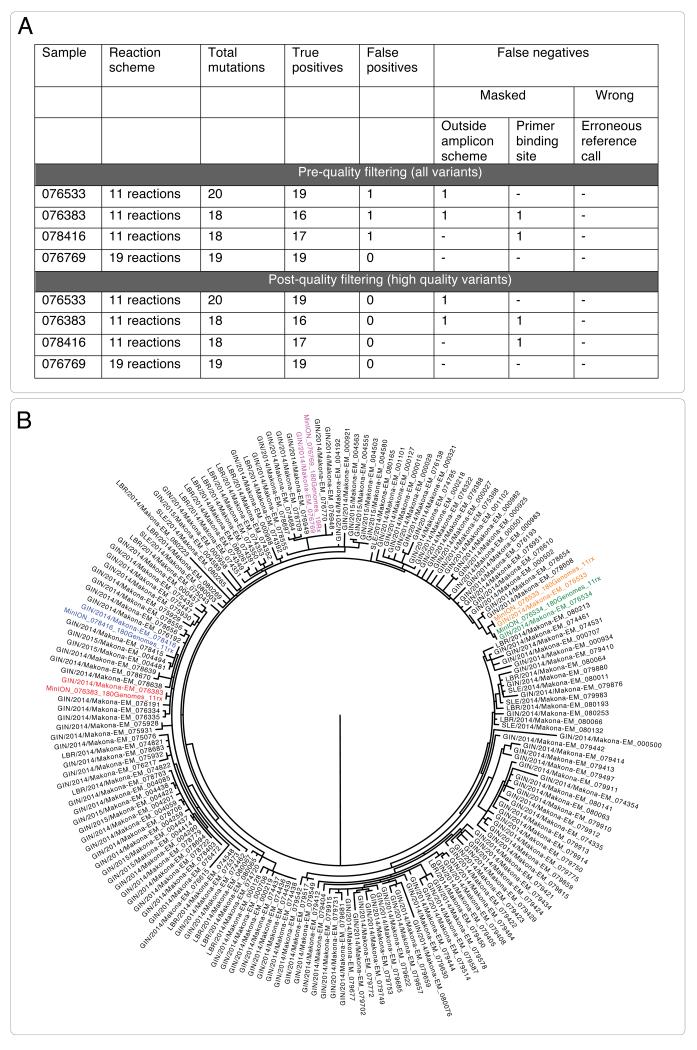

Comment in
-
Ebola: Sequencing on the ground.Nature. 2016 Feb 11;530(7589):167. doi: 10.1038/530167a. Nature. 2016. PMID: 26863977 No abstract available.
-
Portable Nanopore Sequencing for Viral Surveillance.Clin Chem. 2016 Nov;62(11):1427-1429. doi: 10.1373/clinchem.2016.256693. Epub 2016 Sep 16. Clin Chem. 2016. PMID: 27638016 No abstract available.
-
Genomic outbreak surveillance in resource-poor settings.Nat Rev Genet. 2022 Sep;23(9):522-523. doi: 10.1038/s41576-022-00500-w. Nat Rev Genet. 2022. PMID: 35577990 Free PMC article.
References
-
- World Health Organisation [11 November 2015];Ebola Situation Report. 2015 at < http://apps.who.int/ebola/current-situation/ebola-situation-report-11-no...>.
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
