

Late acquisition of mitochondria by a host with chimaeric prokaryotic ancestry
- PMID: 26840490
- PMCID: PMC4780264
- DOI: 10.1038/nature16941
Late acquisition of mitochondria by a host with chimaeric prokaryotic ancestry
Abstract
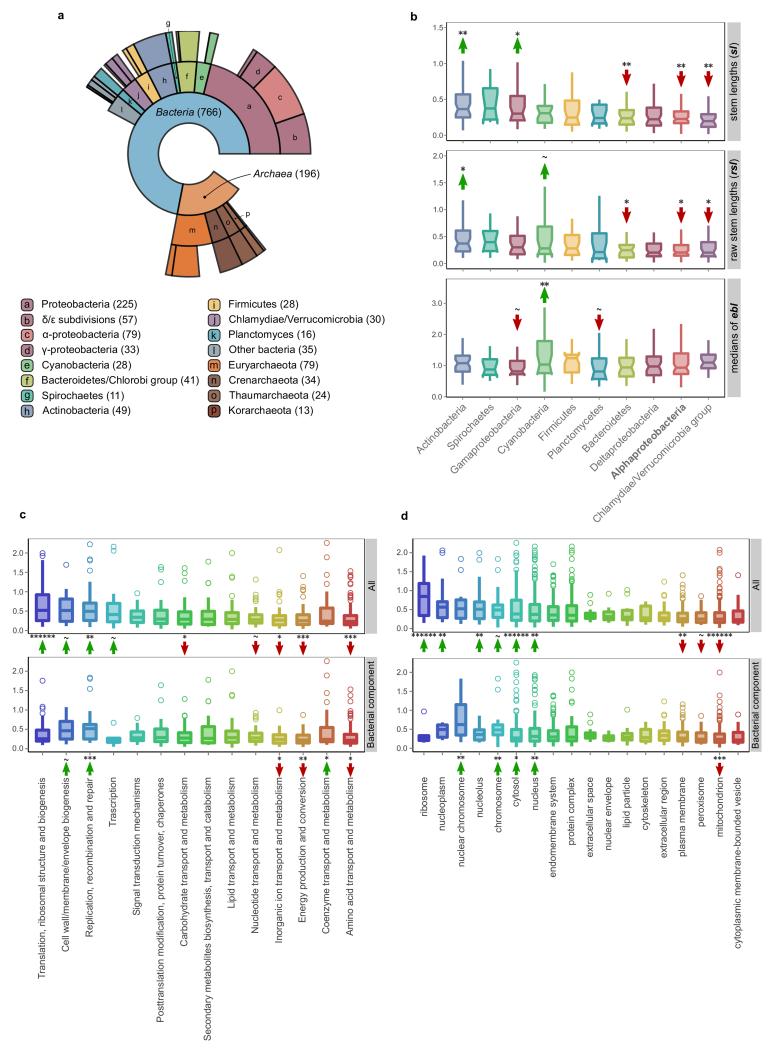
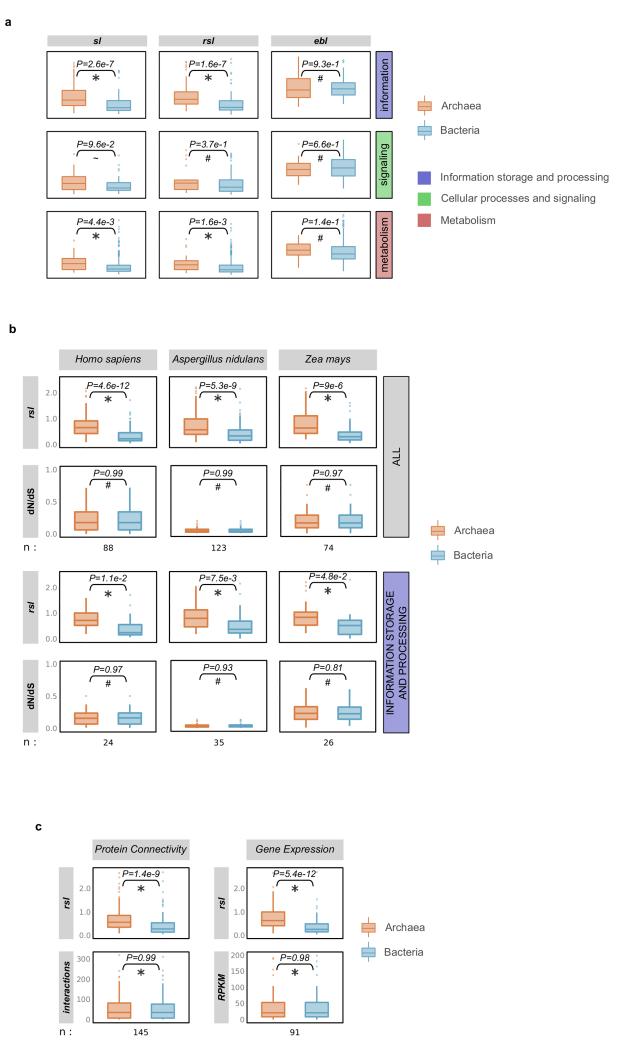
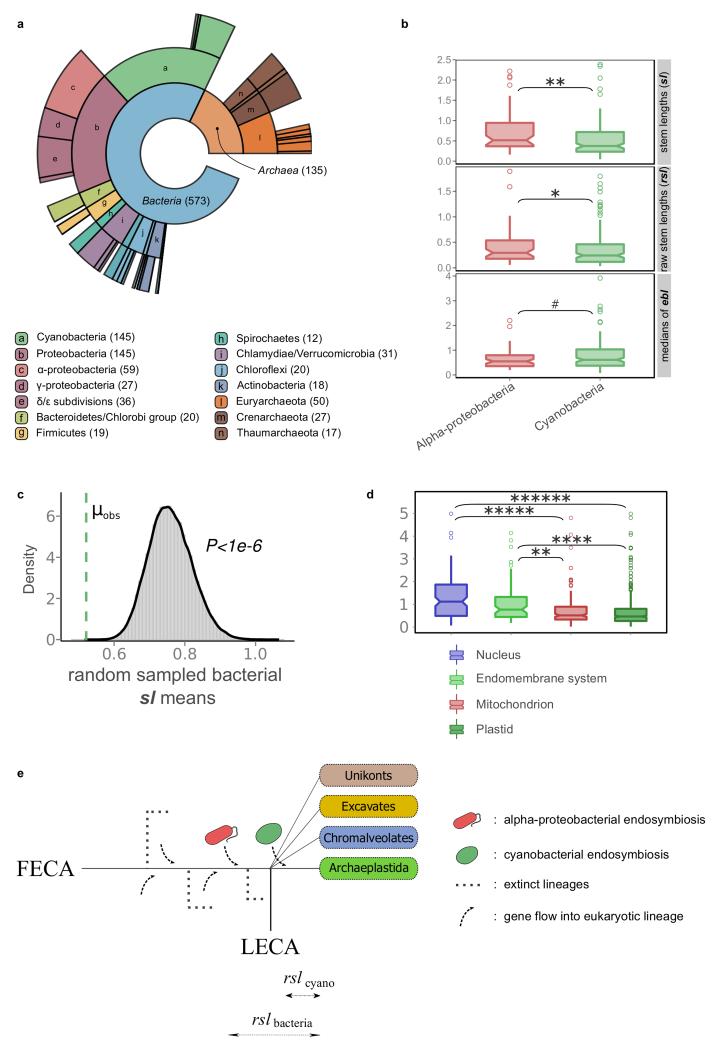
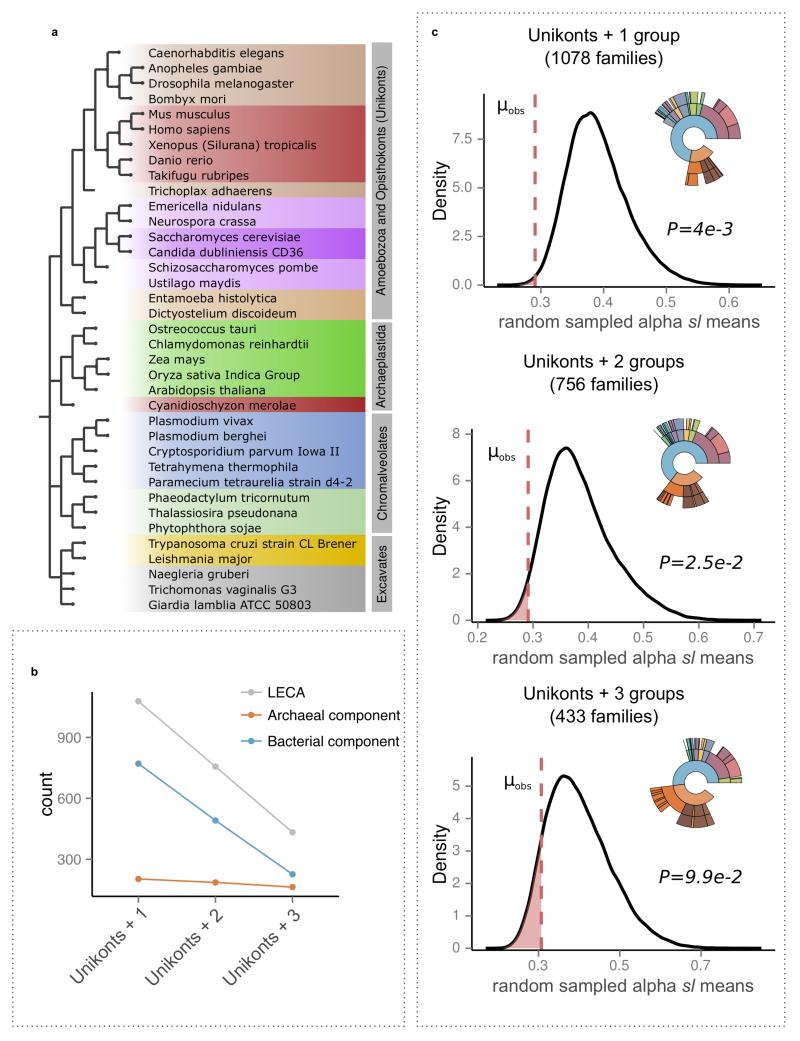
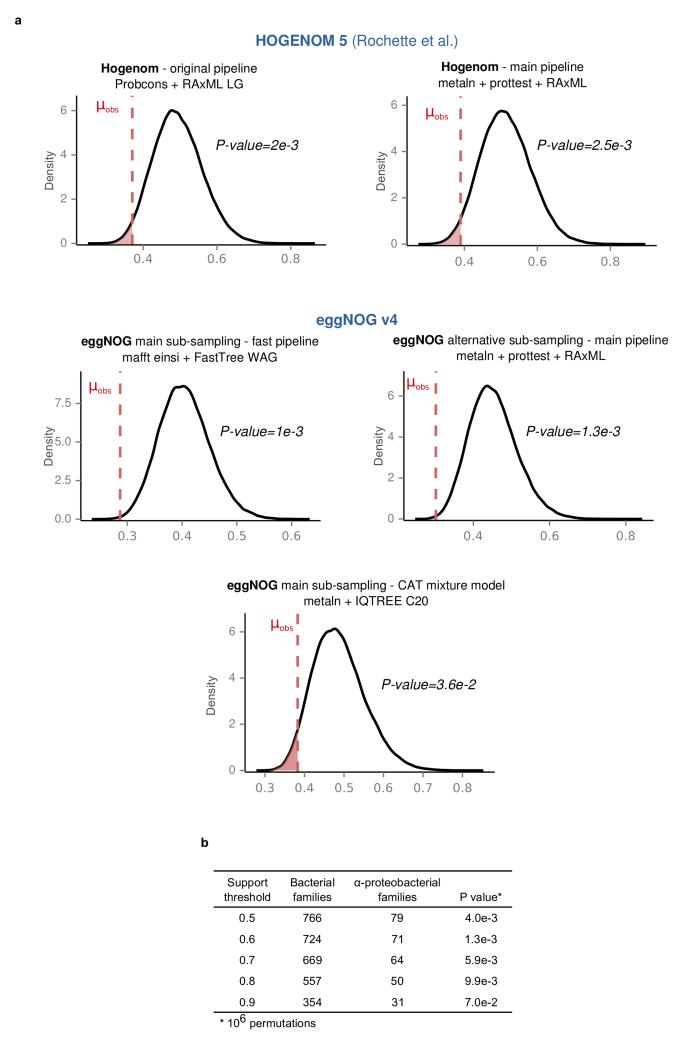
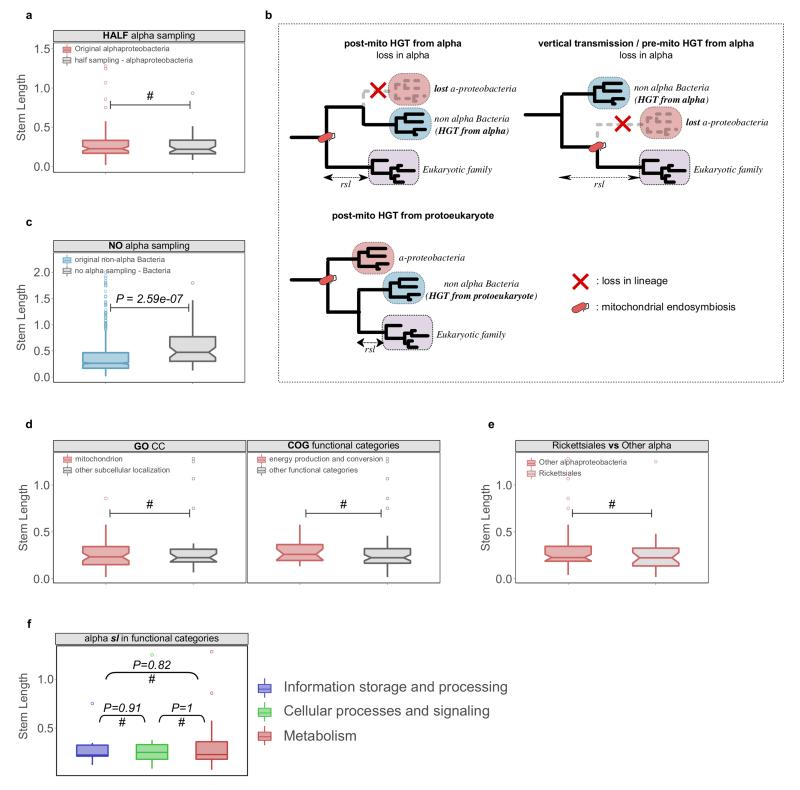
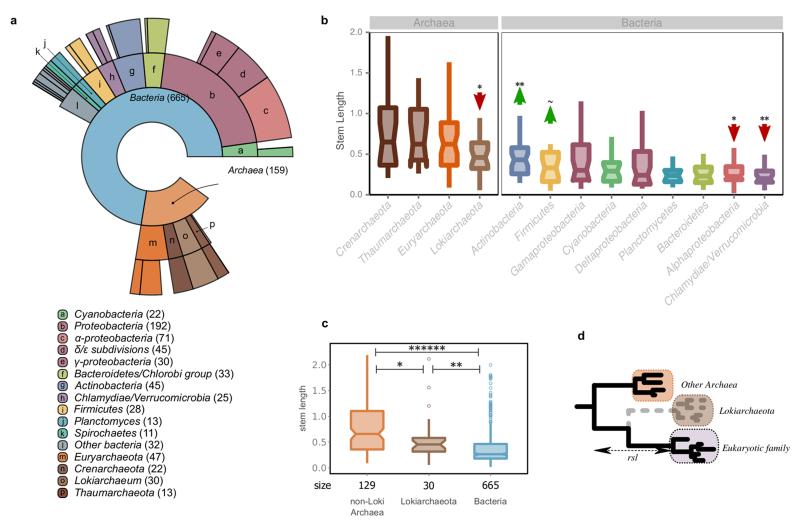
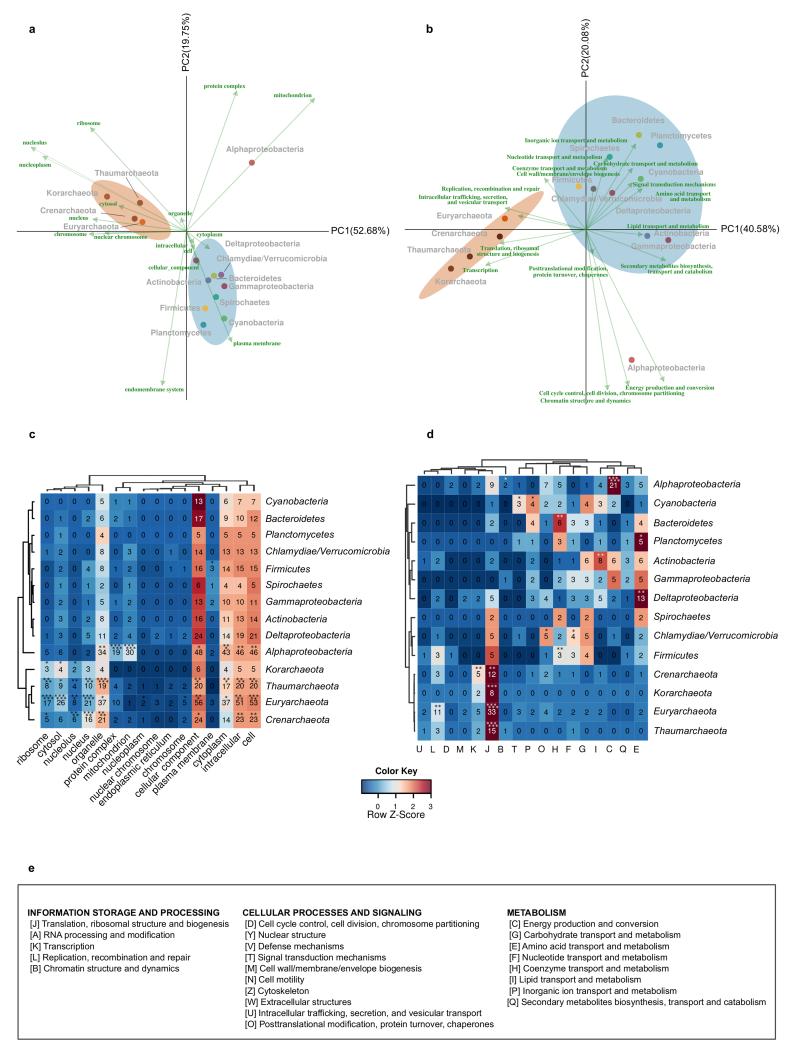
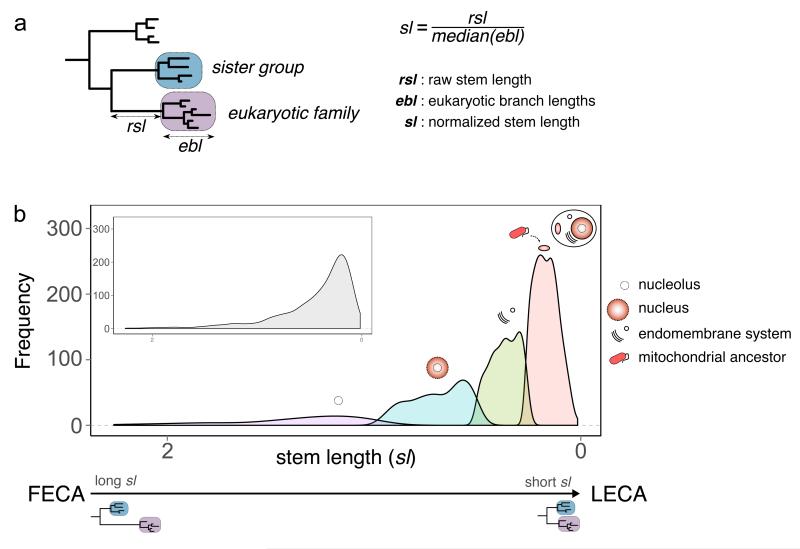
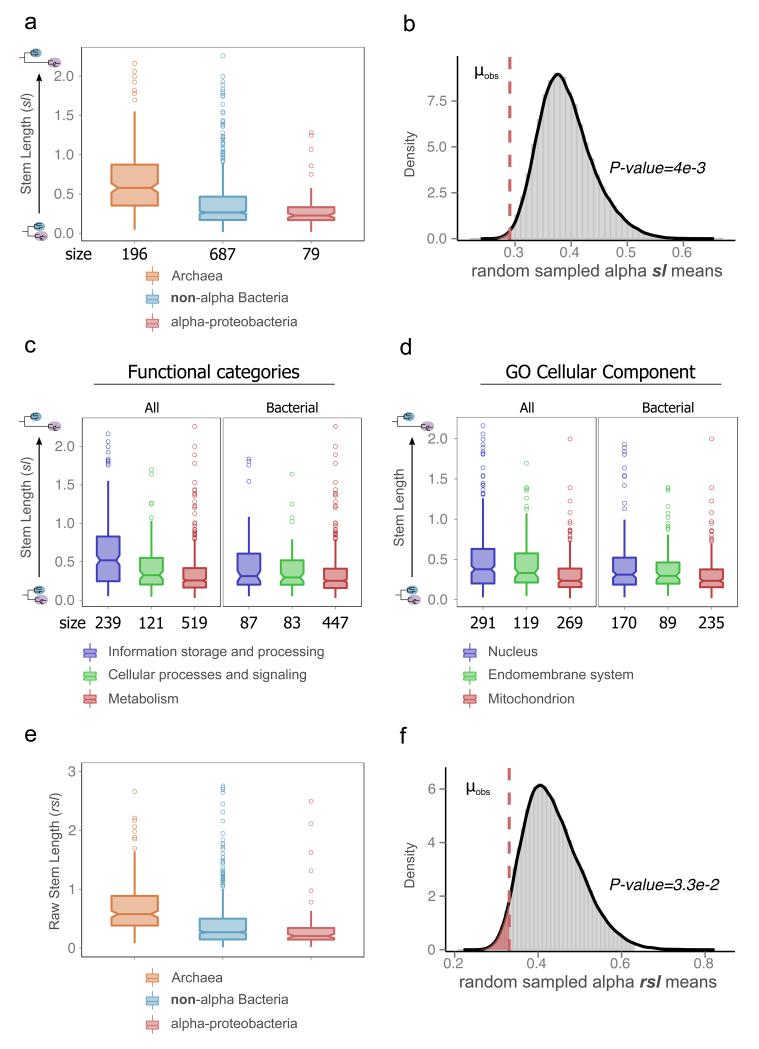
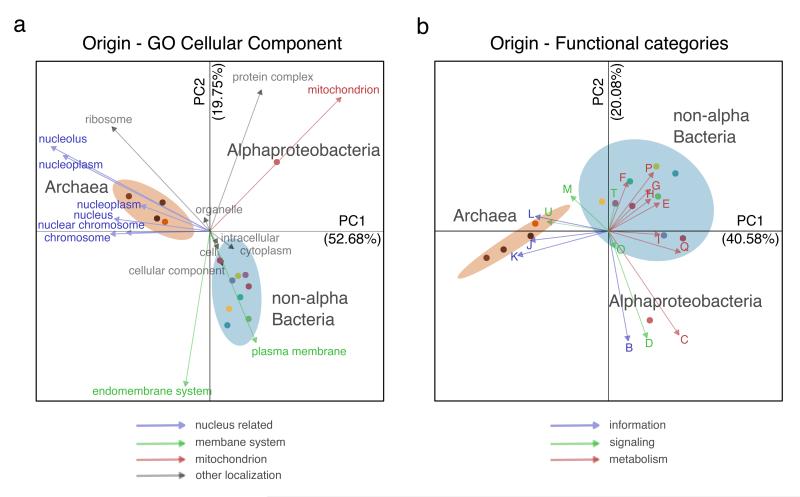
The origin of eukaryotes stands as a major conundrum in biology. Current evidence indicates that the last eukaryotic common ancestor already possessed many eukaryotic hallmarks, including a complex subcellular organization. In addition, the lack of evolutionary intermediates challenges the elucidation of the relative order of emergence of eukaryotic traits. Mitochondria are ubiquitous organelles derived from an alphaproteobacterial endosymbiont. Different hypotheses disagree on whether mitochondria were acquired early or late during eukaryogenesis. Similarly, the nature and complexity of the receiving host are debated, with models ranging from a simple prokaryotic host to an already complex proto-eukaryote. Most competing scenarios can be roughly grouped into either mito-early, which consider the driving force of eukaryogenesis to be mitochondrial endosymbiosis into a simple host, or mito-late, which postulate that a significant complexity predated mitochondrial endosymbiosis. Here we provide evidence for late mitochondrial endosymbiosis. We use phylogenomics to directly test whether proto-mitochondrial proteins were acquired earlier or later than other proteins of the last eukaryotic common ancestor. We find that last eukaryotic common ancestor protein families of alphaproteobacterial ancestry and of mitochondrial localization show the shortest phylogenetic distances to their closest prokaryotic relatives, compared with proteins of different prokaryotic origin or cellular localization. Altogether, our results shed new light on a long-standing question and provide compelling support for the late acquisition of mitochondria into a host that already had a proteome of chimaeric phylogenetic origin. We argue that mitochondrial endosymbiosis was one of the ultimate steps in eukaryogenesis and that it provided the definitive selective advantage to mitochondria-bearing eukaryotes over less complex forms.
Figures
Comment in
-
Evolution: Mitochondria in the second act.Nature. 2016 Mar 3;531(7592):39-40. doi: 10.1038/nature16876. Epub 2016 Feb 3. Nature. 2016. PMID: 26840482 No abstract available.
-
Late Mitochondrial Origin Is an Artifact.Genome Biol Evol. 2017 Feb 1;9(2):373-379. doi: 10.1093/gbe/evx027. Genome Biol Evol. 2017. PMID: 28199635 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
