

Allele-specific inhibitors inactivate mutant KRAS G12C by a trapping mechanism
- PMID: 26841430
- PMCID: PMC4955282
- DOI: 10.1126/science.aad6204
Allele-specific inhibitors inactivate mutant KRAS G12C by a trapping mechanism
Abstract
It is thought that KRAS oncoproteins are constitutively active because their guanosine triphosphatase (GTPase) activity is disabled. Consequently, drugs targeting the inactive or guanosine 5'-diphosphate-bound conformation are not expected to be effective. We describe a mechanism that enables such drugs to inhibit KRAS(G12C) signaling and cancer cell growth. Inhibition requires intact GTPase activity and occurs because drug-bound KRAS(G12C) is insusceptible to nucleotide exchange factors and thus trapped in its inactive state. Indeed, mutants completely lacking GTPase activity and those promoting exchange reduced the potency of the drug. Suppressing nucleotide exchange activity downstream of various tyrosine kinases enhanced KRAS(G12C) inhibition, whereas its potentiation had the opposite effect. These findings reveal that KRAS(G12C) undergoes nucleotide cycling in cancer cells and provide a basis for developing effective therapies to treat KRAS(G12C)-driven cancers.
Copyright © 2016, American Association for the Advancement of Science.
Figures
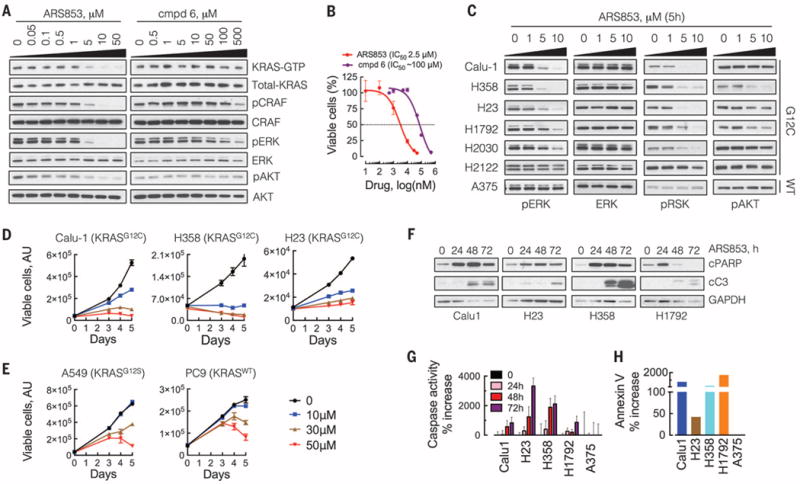
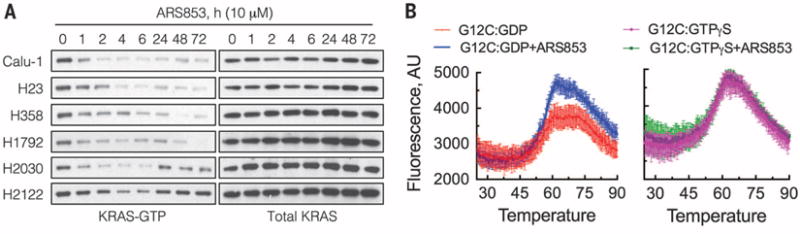
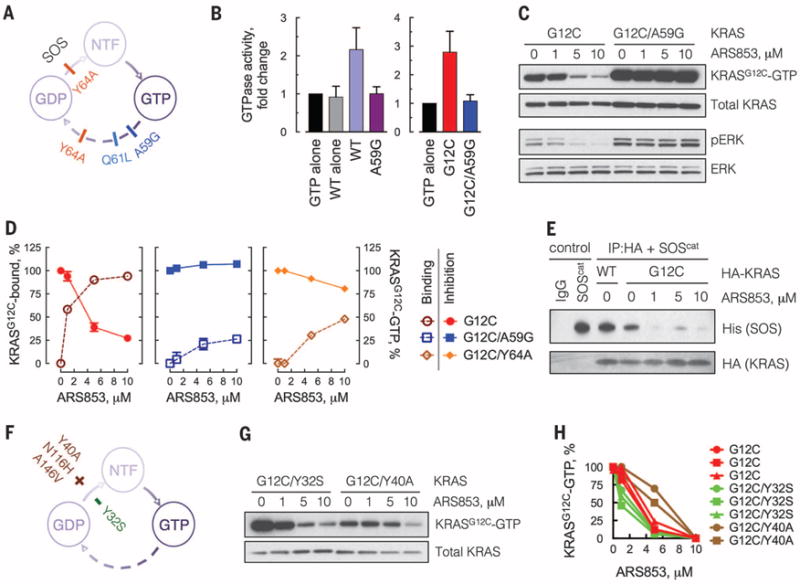
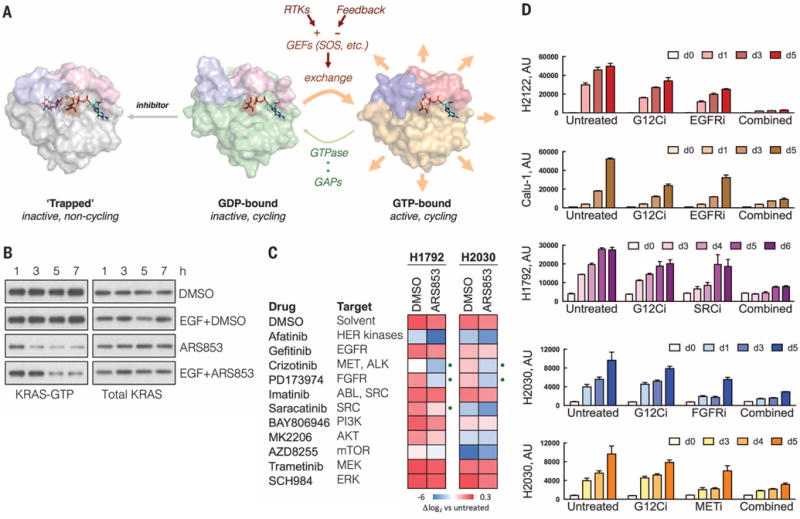
Comment in
-
Signalling: Putting the brakes on KRAS-G12C nucleotide cycling.Nat Rev Cancer. 2016 Mar;16(3):127. doi: 10.1038/nrc.2016.13. Epub 2016 Feb 19. Nat Rev Cancer. 2016. PMID: 26893067 No abstract available.
References
-
- Stephen AG, Esposito D, Bagni RK, McCormick F. Cancer Cell. 2014;25:272–281. - PubMed
-
- Bos JL, Rehmann H, Wittinghofer A. Cell. 2007;129:865–877. - PubMed
-
- Parada LF, Tabin CJ, Shih C, Weinberg RA. Nature. 1982;297:474–478. - PubMed
-
- Santos E, Tronick SR, Aaronson SA, Pulciani S, Barbacid M. Nature. 1982;298:343–347. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
