

Prime, Shock, and Kill: Priming CD4 T Cells from HIV Patients with a BCL-2 Antagonist before HIV Reactivation Reduces HIV Reservoir Size
- PMID: 26842479
- PMCID: PMC4810548
- DOI: 10.1128/JVI.03179-15
Prime, Shock, and Kill: Priming CD4 T Cells from HIV Patients with a BCL-2 Antagonist before HIV Reactivation Reduces HIV Reservoir Size
Abstract
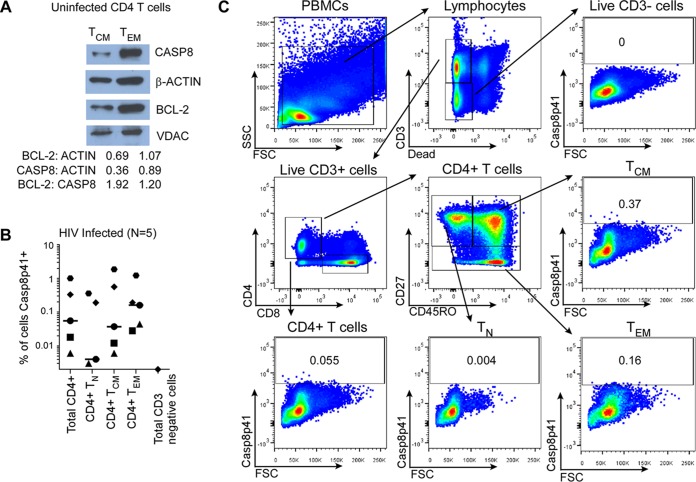
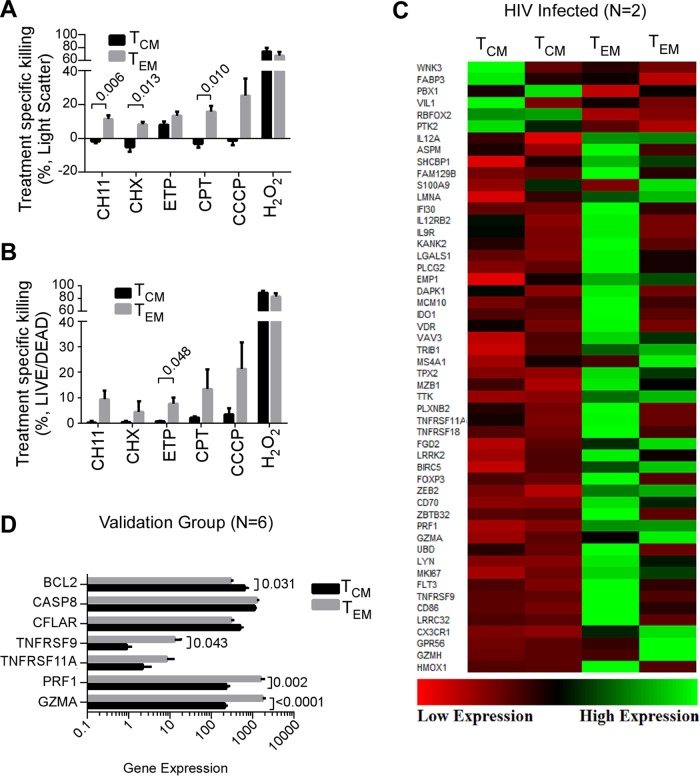
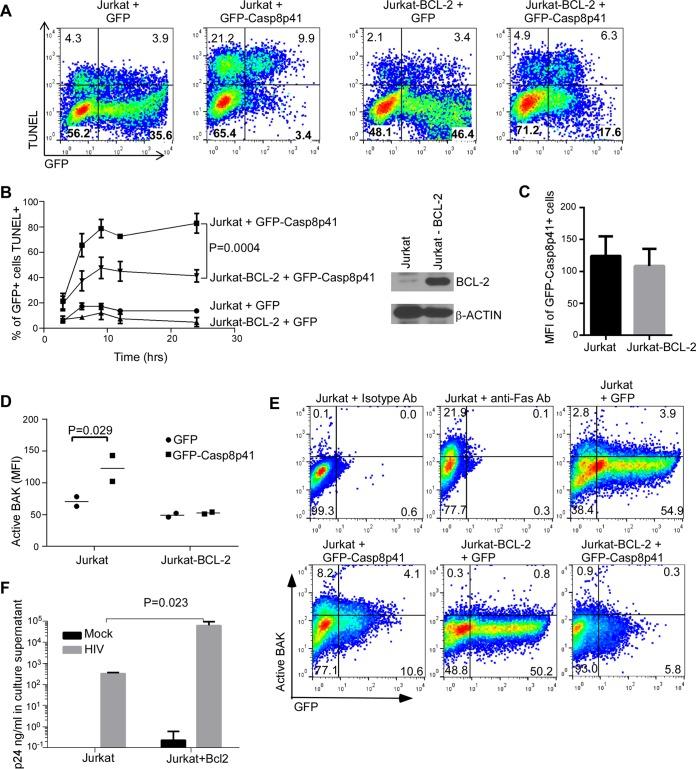
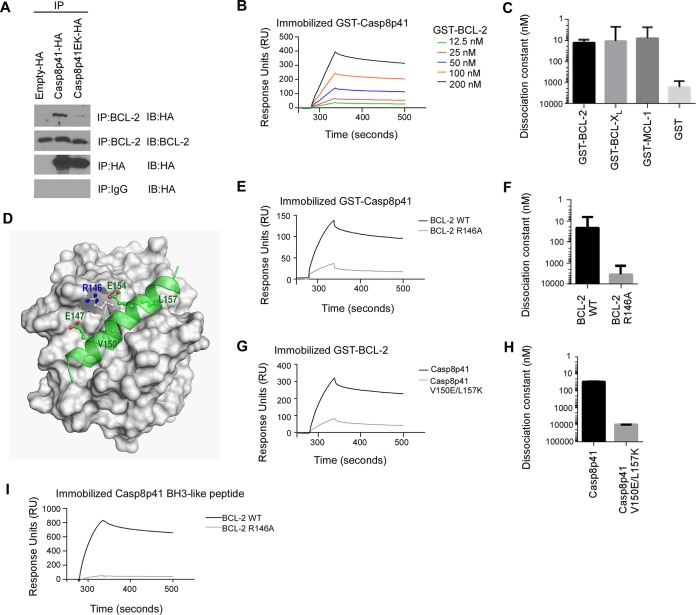
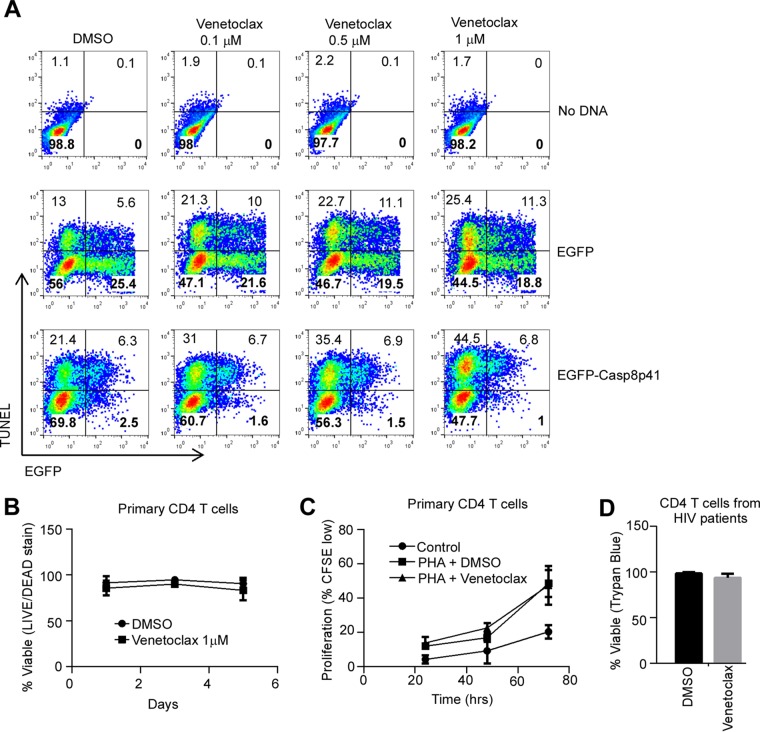
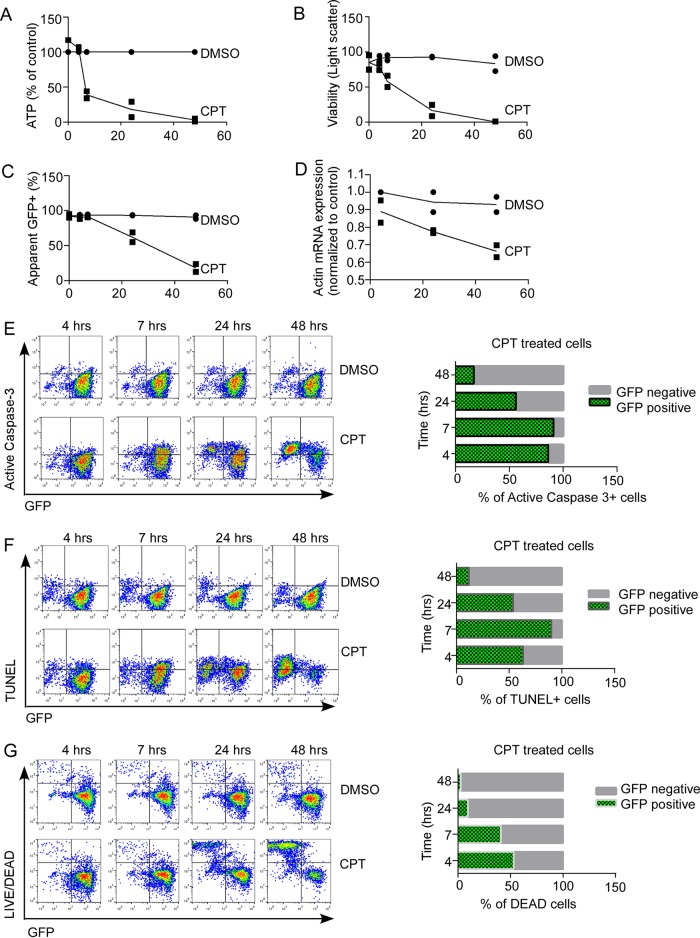
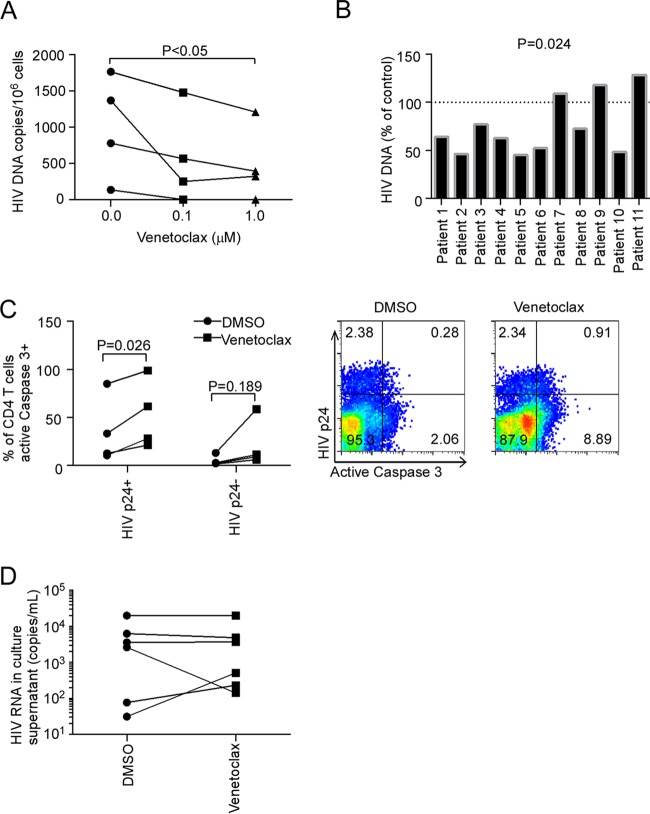
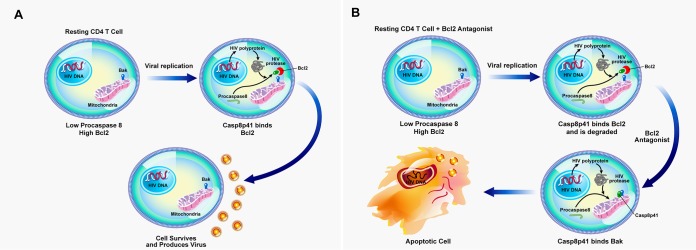
Understanding how some HIV-infected cells resist the cytotoxicity of HIV replication is crucial to enabling HIV cure efforts. HIV killing of CD4 T cells that replicate HIV can involve HIV protease-mediated cleavage of procaspase 8 to generate a fragment (Casp8p41) that directly binds and activates the mitochondrial proapoptotic protein BAK. Here, we demonstrate that Casp8p41 also binds with nanomolar affinity to the antiapoptotic protein Bcl-2, which sequesters Casp8p41 and prevents apoptosis. Further, we show that central memory CD4 T cells (TCM) from HIV-infected individuals have heightened expression of BCL-2 relative to procaspase 8, possibly explaining the persistence of HIV-infected TCMdespite generation of Casp8p41. Consistent with this hypothesis, the selective BCL-2 antagonist venetoclax induced minimal killing of uninfected CD4 T cells but markedly increased the death of CD4 T cells and diminished cell-associated HIV DNA when CD4 T cells from antiretroviral therapy (ART)-suppressed HIV patients were induced with αCD3/αCD28 to reactivate HIVex vivo Thus, priming CD4 T cells from ART suppressed HIV patients with a BCL-2 antagonist, followed by HIV reactivation, achieves reductions in cell-associated HIV DNA, whereas HIV reactivation alone does not.
Importance: HIV infection is incurable due to a long-lived reservoir of HIV(+)memory CD4 T cells, and no clinically relevant interventions have been identified that reduce the number of these HIV DNA-containing cells. Since postintegration HIV replication can result in HIV protease generation of Casp8p41, which activates BAK, causing infected CD4 T cell death, we sought to determine whether this occurs in memory CD4 T cells. Here, we demonstrate that memory CD4 T cells can generate Casp8p41 and yet are intrinsically resistant to death induced by diverse stimuli, including Casp8p41. Furthermore, BCL-2 expression is relatively increased in these cells and directly binds and inhibits Casp8p41's proapoptotic effects. Antagonizing BCL-2 with venetoclax derepresses this antagonism, resulting in death, preferentially in HIV DNA containing cells, since only these cells generate Casp8p41. Thus, BCL-2 antagonism is a clinically relevant intervention with the potential to reduce HIV reservoir size in patients.
Copyright © 2016, American Society for Microbiology. All Rights Reserved.
Figures
Similar articles
-
Maintenance of the HIV Reservoir Is Antagonized by Selective BCL2 Inhibition.J Virol. 2017 May 12;91(11):e00012-17. doi: 10.1128/JVI.00012-17. Print 2017 Jun 1. J Virol. 2017. PMID: 28331083 Free PMC article.
-
The HIV-1-specific protein Casp8p41 induces death of infected cells through Bax/Bak.J Virol. 2011 Aug;85(16):7965-75. doi: 10.1128/JVI.02515-10. Epub 2011 Jun 8. J Virol. 2011. PMID: 21653671 Free PMC article.
-
Increasing procaspase 8 expression using repurposed drugs to induce HIV infected cell death in ex vivo patient cells.PLoS One. 2017 Jun 19;12(6):e0179327. doi: 10.1371/journal.pone.0179327. eCollection 2017. PLoS One. 2017. PMID: 28628632 Free PMC article.
-
Casp8p41: The Protean Mediator of Death in CD4 T-cells that Replicate HIV.J Cell Death. 2016 Sep 27;9:9-17. doi: 10.4137/JCD.S39872. eCollection 2016. J Cell Death. 2016. PMID: 27721655 Free PMC article. Review.
-
Blocking Formation of the Stable HIV Reservoir: A New Perspective for HIV-1 Cure.Front Immunol. 2019 Aug 22;10:1966. doi: 10.3389/fimmu.2019.01966. eCollection 2019. Front Immunol. 2019. PMID: 31507594 Free PMC article. Review.
Cited by
-
Antiretroviral treatment, government policy and economy of HIV/AIDS in Brazil: is it time for HIV cure in the country?AIDS Res Ther. 2019 Aug 14;16(1):19. doi: 10.1186/s12981-019-0234-2. AIDS Res Ther. 2019. PMID: 31412889 Free PMC article. Review.
-
Highlights from the Fourth Biennial Strategies for an HIV Cure Meeting, 10-12 October 2018, Bethesda, MD, USA.J Virus Erad. 2019 Jan 1;5(1):50-59. doi: 10.1016/S2055-6640(20)30280-6. J Virus Erad. 2019. PMID: 30800428 Free PMC article.
-
Microglia and macrophages alterations in the CNS during acute SIV infection: a single-cell analysis in rhesus macaques.bioRxiv [Preprint]. 2024 Apr 4:2024.04.04.588047. doi: 10.1101/2024.04.04.588047. bioRxiv. 2024. Update in: PLoS Pathog. 2024 Sep 16;20(9):e1012168. doi: 10.1371/journal.ppat.1012168. PMID: 38617282 Free PMC article. Updated. Preprint.
-
Small molecule inhibitors of transcriptional cyclin-dependent kinases impose HIV-1 latency, presenting "block and lock" treatment strategies.Antimicrob Agents Chemother. 2024 Mar 6;68(3):e0107223. doi: 10.1128/aac.01072-23. Epub 2024 Feb 6. Antimicrob Agents Chemother. 2024. PMID: 38319085 Free PMC article.
-
Immune-mediated strategies to solving the HIV reservoir problem.Nat Rev Immunol. 2025 Jul;25(7):542-553. doi: 10.1038/s41577-025-01136-7. Epub 2025 Feb 13. Nat Rev Immunol. 2025. PMID: 39948261 Review.
References
-
- Archin NM, Liberty AL, Kashuba AD, Choudhary SK, Kuruc JD, Crooks AM, Parker DC, Anderson EM, Kearney MF, Strain MC, Richman DD, Hudgens MG, Bosch RJ, Coffin JM, Eron JJ, Hazuda DJ, Margolis DM. 2012. Administration of vorinostat disrupts HIV-1 latency in patients on antiretroviral therapy. Nature 487:482–485. doi: 10.1038/nature11286. - DOI - PMC - PubMed
-
- Elliott JH, Wightman F, Solomon A, Ghneim K, Ahlers J, Cameron MJ, Smith MZ, Spelman T, McMahon J, Velayudham P, Brown G, Roney J, Watson J, Prince MH, Hoy JF, Chomont N, Fromentin R, Procopio FA, Zeidan J, Palmer S, Odevall L, Johnstone RW, Martin BP, Sinclair E, Deeks SG, Hazuda DJ, Cameron PU, Sekaly RP, Lewin SR. 2014. Activation of HIV transcription with short-course vorinostat in HIV-infected patients on suppressive antiretroviral therapy. PLoS Pathog 10:e1004473. doi: 10.1371/journal.ppat.1004473. - DOI - PMC - PubMed
-
- Rasmussen TA, Tolstrup M, Brinkmann CR, Olesen R, Erikstrup C, Solomon A, Winckelmann A, Palmer S, Dinarello C, Buzon M, Lichterfeld M, Lewin SR, Løstergaard-Søgaard OS. Panobinostat, a histone deacetylase inhibitor, for latent-virus reactivation in HIV-infected patients on suppressive antiretroviral therapy: a phase 1/2, single group, clinical trial. Lancet HIV 1:e13–e21. doi: 10.1016/S2352-3018(14)70014-1. - DOI - PubMed
-
- Sogaard OS, Graversen ME, Leth S, Olesen R, Brinkmann CR, Nissen SK, Kjaer AS, Schleimann MH, Denton PW, Hey-Cunningham WJ, Koelsch KK, Pantaleo G, Krogsgaard K, Sommerfelt M, Fromentin R, Chomont N, Rasmussen TA, Ostergaard L, Tolstrup M. 2015. The depsipeptide romidepsin reverses HIV-1 latency in vivo. PLoS Pathog 11:e1005142. doi: 10.1371/journal.ppat.1005142. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
