

Differential contribution of genomic regions to marked genetic variation and prediction of quantitative traits in broiler chickens
- PMID: 26842494
- PMCID: PMC4739338
- DOI: 10.1186/s12711-016-0187-z
Differential contribution of genomic regions to marked genetic variation and prediction of quantitative traits in broiler chickens
Abstract
Background: Genome-wide association studies in humans have found enrichment of trait-associated single nucleotide polymorphisms (SNPs) in coding regions of the genome and depletion of these in intergenic regions. However, a recent release of the ENCyclopedia of DNA elements showed that ~80 % of the human genome has a biochemical function. Similar studies on the chicken genome are lacking, thus assessing the relative contribution of its genic and non-genic regions to variation is relevant for biological studies and genetic improvement of chicken populations.
Methods: A dataset including 1351 birds that were genotyped with the 600K Affymetrix platform was used. We partitioned SNPs according to genome annotation data into six classes to characterize the relative contribution of genic and non-genic regions to genetic variation as well as their predictive power using all available quality-filtered SNPs. Target traits were body weight, ultrasound measurement of breast muscle and hen house egg production in broiler chickens. Six genomic regions were considered: intergenic regions, introns, missense, synonymous, 5' and 3' untranslated regions, and regions that are located 5 kb upstream and downstream of coding genes. Genomic relationship matrices were constructed for each genomic region and fitted in the models, separately or simultaneously. Kernel-based ridge regression was used to estimate variance components and assess predictive ability. Contribution of each class of genomic regions to dominance variance was also considered.
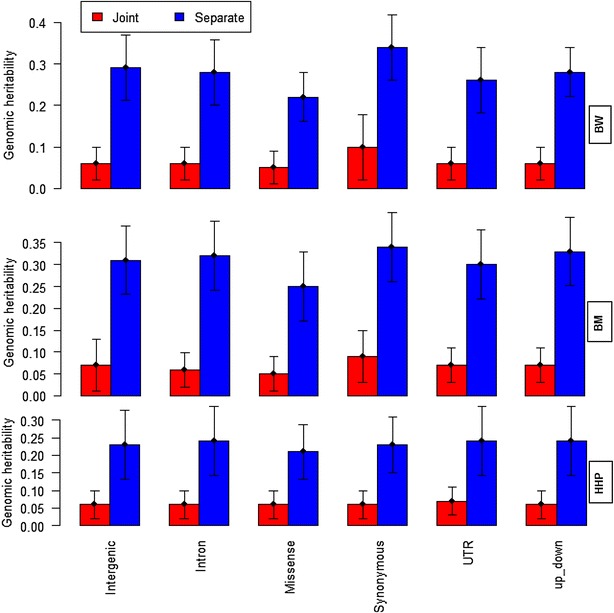
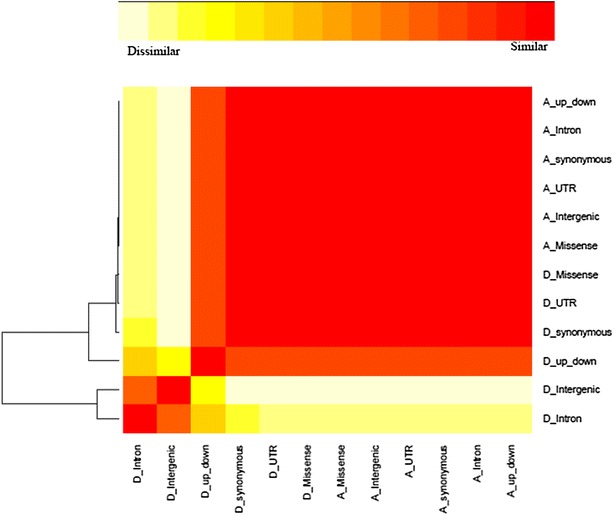
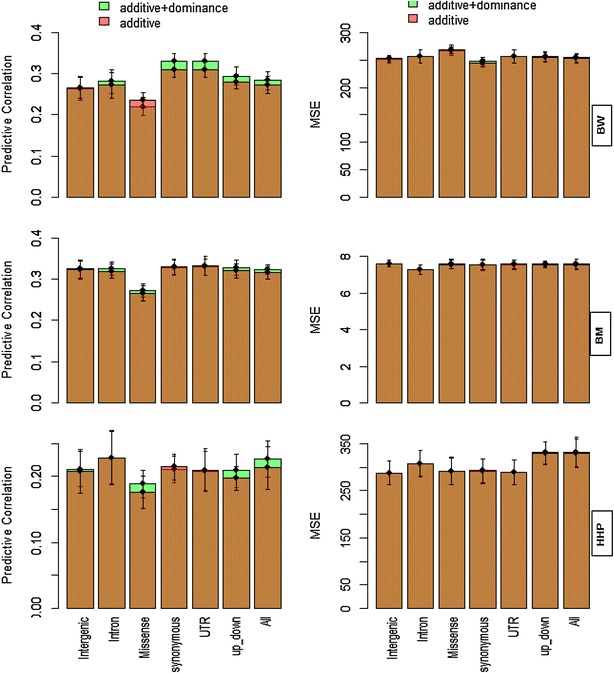
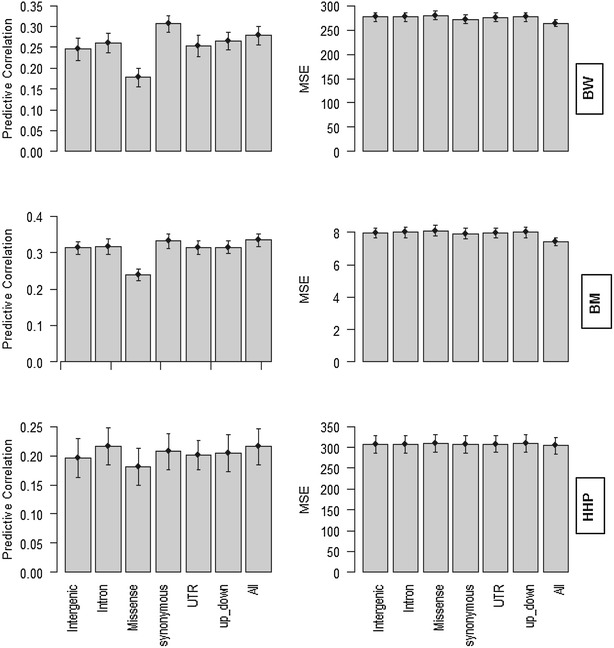
Results: Variance component estimates indicated that all genomic regions contributed to marked additive genetic variation and that the class of synonymous regions tended to have the greatest contribution. The marked dominance genetic variation explained by each class of genomic regions was similar and negligible (~0.05). In terms of prediction mean-square error, the whole-genome approach showed the best predictive ability.
Conclusions: All genic and non-genic regions contributed to phenotypic variation for the three traits studied. Overall, the contribution of additive genetic variance to the total genetic variance was much greater than that of dominance variance. Our results show that all genomic regions are important for the prediction of the targeted traits, and the whole-genome approach was reaffirmed as the best tool for genome-enabled prediction of quantitative traits.
Figures
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
