

Models of human core transcriptional regulatory circuitries
- PMID: 26843070
- PMCID: PMC4772020
- DOI: 10.1101/gr.197590.115
Models of human core transcriptional regulatory circuitries
Abstract
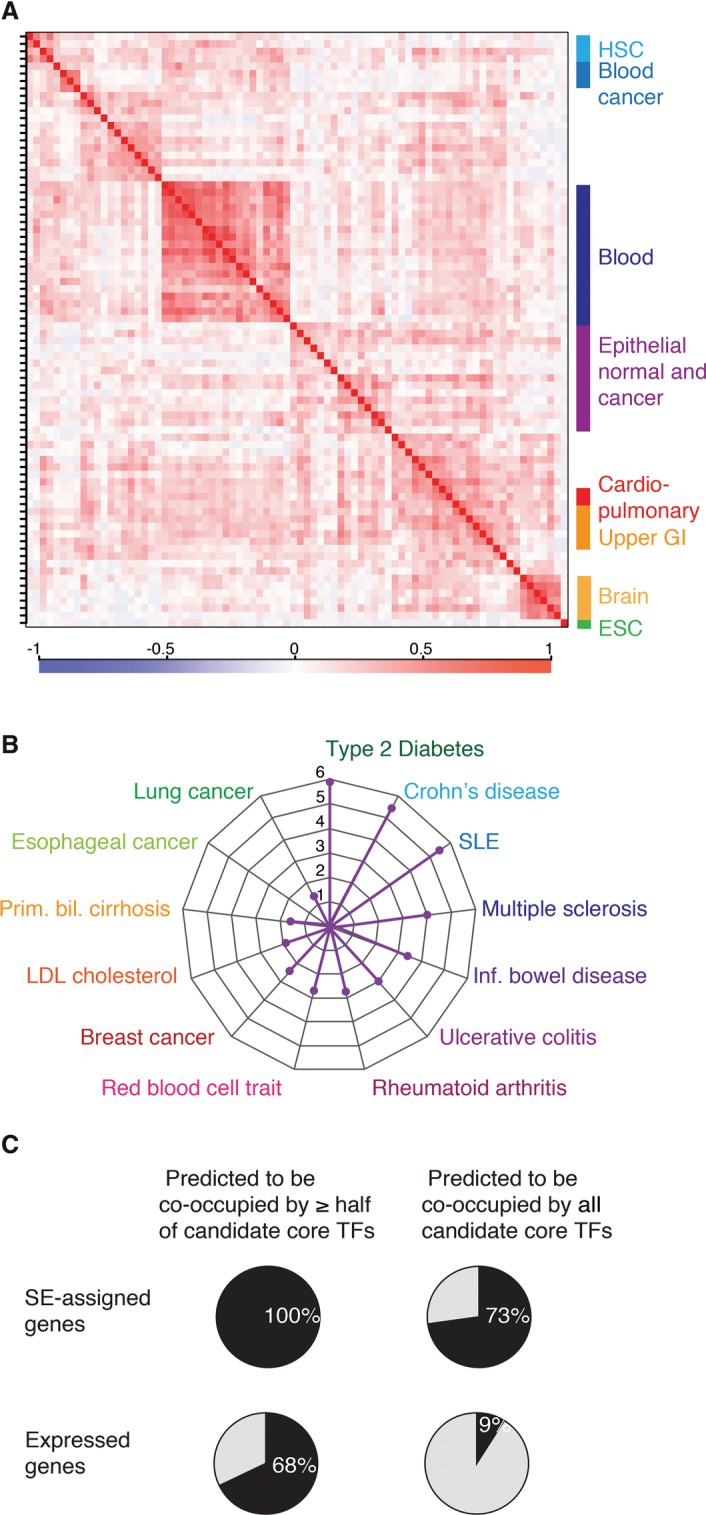
A small set of core transcription factors (TFs) dominates control of the gene expression program in embryonic stem cells and other well-studied cellular models. These core TFs collectively regulate their own gene expression, thus forming an interconnected auto-regulatory loop that can be considered the core transcriptional regulatory circuitry (CRC) for that cell type. There is limited knowledge of core TFs, and thus models of core regulatory circuitry, for most cell types. We recently discovered that genes encoding known core TFs forming CRCs are driven by super-enhancers, which provides an opportunity to systematically predict CRCs in poorly studied cell types through super-enhancer mapping. Here, we use super-enhancer maps to generate CRC models for 75 human cell and tissue types. These core circuitry models should prove valuable for further investigating cell-type-specific transcriptional regulation in healthy and diseased cells.
© 2016 Saint-André et al.; Published by Cold Spring Harbor Laboratory Press.
Figures
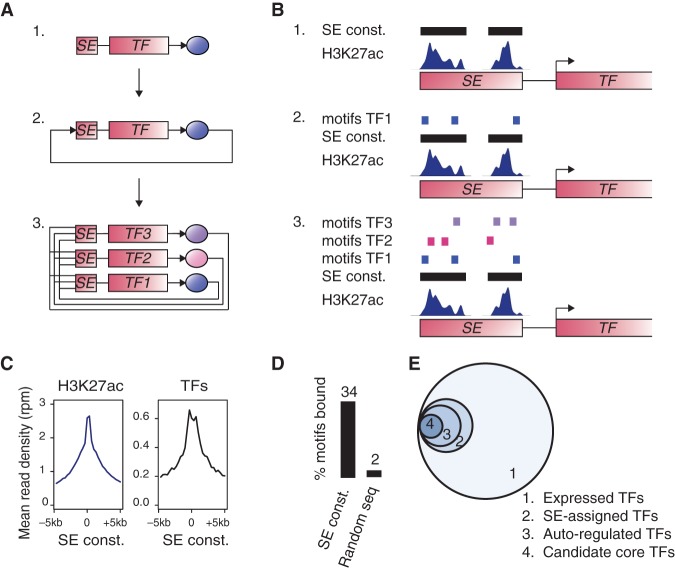
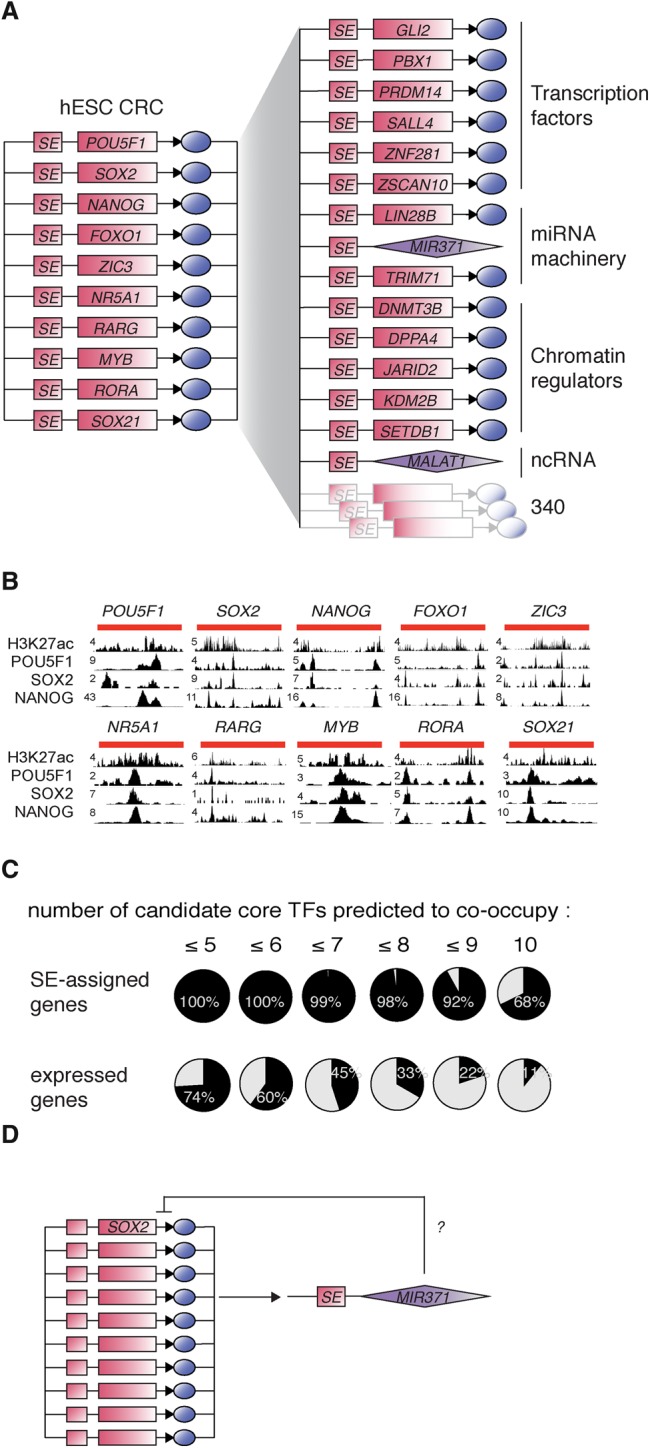

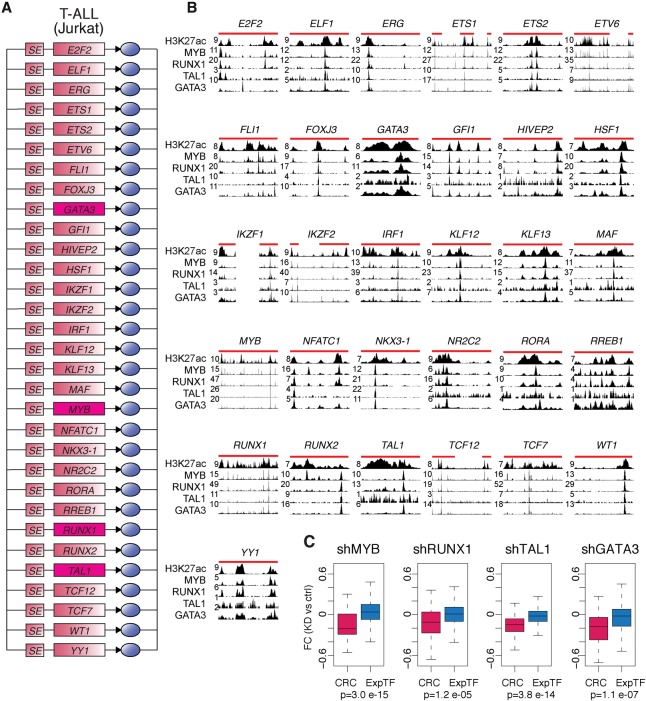
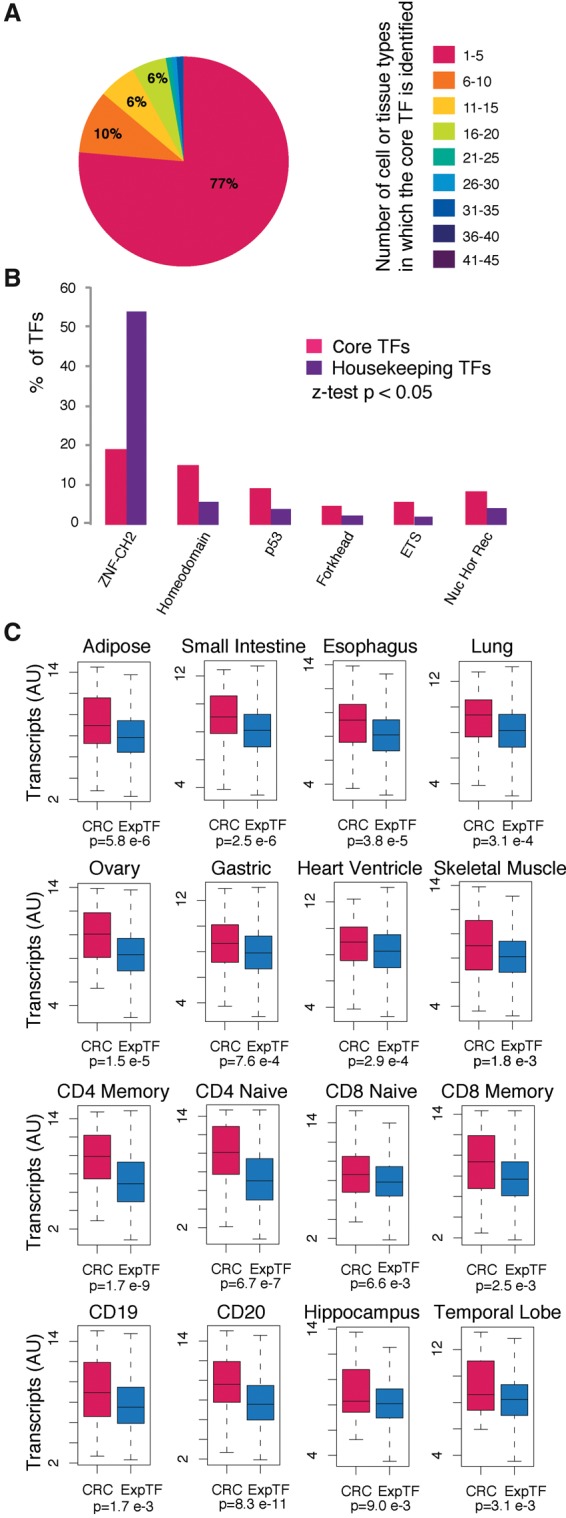
References
-
- Alon U. 2007. Network motifs: theory and experimental approaches. Nat Rev Genet 8: 450–461. - PubMed
-
- Bar-Joseph Z, Gerber GK, Lee TI, Rinaldi NJ, Yoo JY, Robert F, Gordon DB, Fraenkel E, Jaakkola TS, Young RA, et al. 2003. Computational discovery of gene modules and regulatory networks. Nat Biotechnol 21: 1337–1342. - PubMed
-
- Barnea E, Bergman Y. 2000. Synergy of SF1 and RAR in activation of Oct-3/4 promoter. J Biol Chem 275: 6608–6619. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases