

Cross-platform normalization of microarray and RNA-seq data for machine learning applications
- PMID: 26844019
- PMCID: PMC4736986
- DOI: 10.7717/peerj.1621
Cross-platform normalization of microarray and RNA-seq data for machine learning applications
Abstract
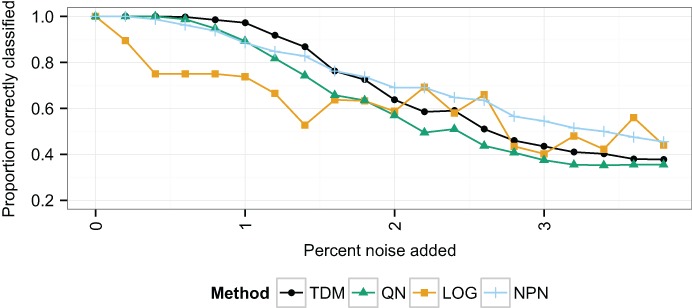
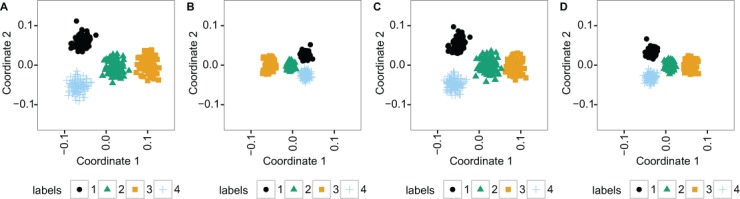
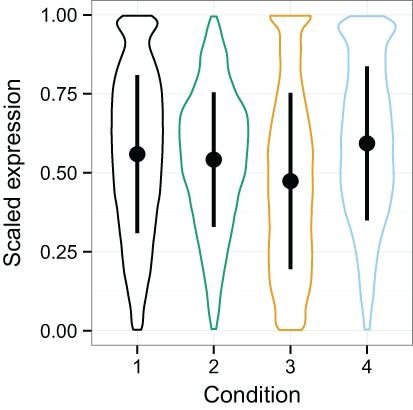
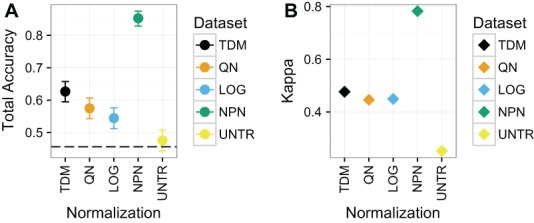
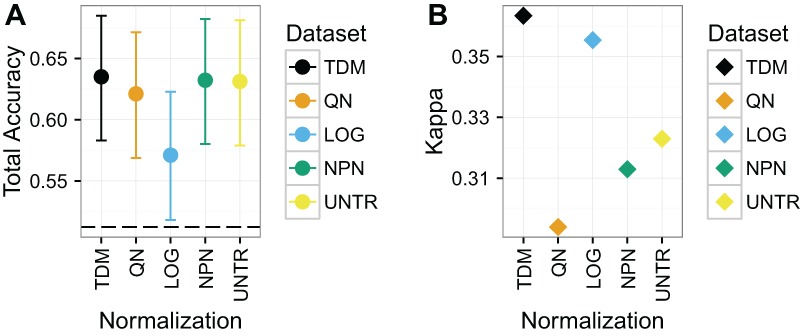
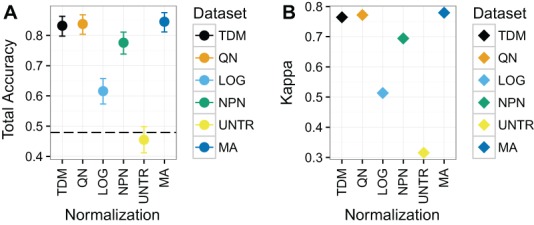
Large, publicly available gene expression datasets are often analyzed with the aid of machine learning algorithms. Although RNA-seq is increasingly the technology of choice, a wealth of expression data already exist in the form of microarray data. If machine learning models built from legacy data can be applied to RNA-seq data, larger, more diverse training datasets can be created and validation can be performed on newly generated data. We developed Training Distribution Matching (TDM), which transforms RNA-seq data for use with models constructed from legacy platforms. We evaluated TDM, as well as quantile normalization, nonparanormal transformation, and a simple log 2 transformation, on both simulated and biological datasets of gene expression. Our evaluation included both supervised and unsupervised machine learning approaches. We found that TDM exhibited consistently strong performance across settings and that quantile normalization also performed well in many circumstances. We also provide a TDM package for the R programming language.
Keywords: Cross-platform normalization; Distribution; Gene expression; Machine learning; Microarray; Nonparanormal transformation; Normalization; Quantile normalization; RNA-sequencing; Training.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
References
-
- Atak ZK, Gianfelici V, Hulselmans G, De Keersmaecker K, Devasia AG, Geerdens E, Mentens N, Chiaretti S, Durinck K, Uyttebroeck A, Vandenberghe P, Wlodarska I, Cloos J, Foà R, Speleman F, Cools J, Aerts S. Comprehensive analysis of transcriptome variation uncovers known and novel driver events in t-cell acute lymphoblastic leukemia. PLoS Genetics. 2013;9(12):e1621. doi: 10.1371/journal.pgen.1003997. - DOI - PMC - PubMed
-
- Bolstad BM. Preprocesscore: A Collection of Pre-Processing Functions. (R package version 1.30.0) 2015
-
- Curtis C, Shah SP, Chin S-F, Turashvili G, Rueda OM, Dunning MJ, Speed D, Lynch AG, Samarajiwa S, Yuan Y, Gräf S, Ha G, Haffari G, Bashashati A, Russell R, McKinney S, METABRIC Group. Langerød A, Green A, Provenzano E, Wishart G, Pinder S, Watson P, Markowetz F, Murphy L, Ellis I, Purushotham A, Børresen-Dale AL-L, Brenton JD, Tavaré S, Caldas C, Aparicio S. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature. 2012;486(7403):346–352. doi: 10.1038/nature10983. - DOI - PMC - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
