

Review
doi: 10.1016/j.neuron.2015.12.023.
Axon Self-Destruction: New Links among SARM1, MAPKs, and NAD+ Metabolism
Affiliations
- PMID: 26844829
- PMCID: PMC4742785
- DOI: 10.1016/j.neuron.2015.12.023
Item in Clipboard
Review
Axon Self-Destruction: New Links among SARM1, MAPKs, and NAD+ Metabolism
Neuron.
.
Abstract
Wallerian axon degeneration is a form of programmed subcellular death that promotes axon breakdown in disease and injury. Active degeneration requires SARM1 and MAP kinases, including DLK, while the NAD+ synthetic enzyme NMNAT2 prevents degeneration. New studies reveal that these pathways cooperate in a locally mediated axon destruction program, with NAD+ metabolism playing a central role. Here, we review the biology of Wallerian-type axon degeneration and discuss the most recent findings, with special emphasis on critical signaling events and their potential as therapeutic targets for axonopathy.
Copyright © 2016 Elsevier Inc. All rights reserved.
Figures
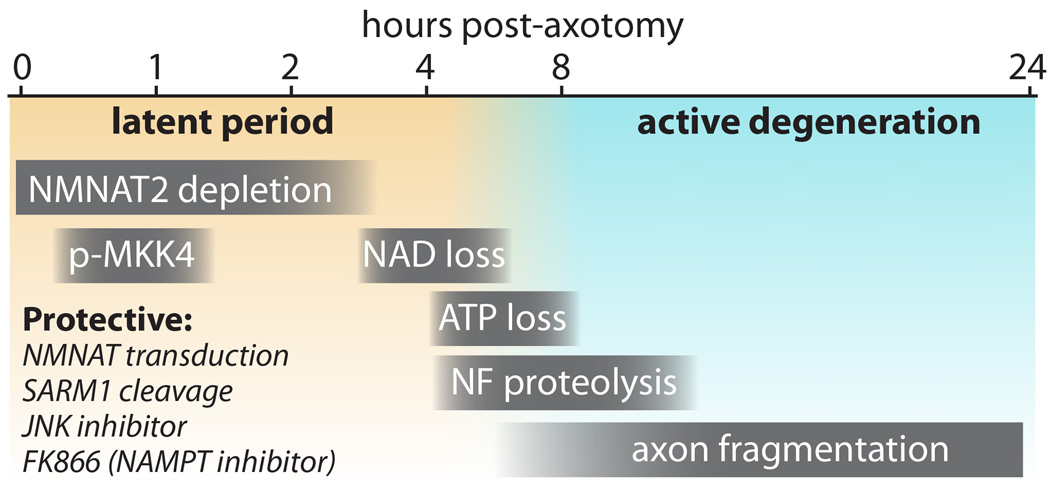
Time course of events in axon degeneration in cultured DRG neurons. The “latent period” (from injury to ~4–6 hours) precedes morphological changes and is characterized by NMNAT2 depletion (Di Stefano et al., 2015), and transient phosphorylation of MKK4 at S257/T261 (Yang et al., 2015). During this window, axon degeneration can be halted by several protective manipulations including NMNAT protein transduction (Sasaki and Milbrandt, 2010), SARM1 cleavage (Gerdts et al., 2015), and addition of FK866 (Sasaki et al., 2009b; Di Stefano et al., 2015) or JNK inhibitors (Miller et al., 2009). As NAD+ declines from 3–6 hours (Wang et al., 2005), ATP levels also decline (Yang et al., 2015), and an active and irreversible phase of axon degeneration begins with neurofilament (NF) proteolysis (Yang et al., 2013) stimulated by calcium influx and finally frank morphologic fragmentation of the axons. These events also occur in vivo, albeit over a slower time course.
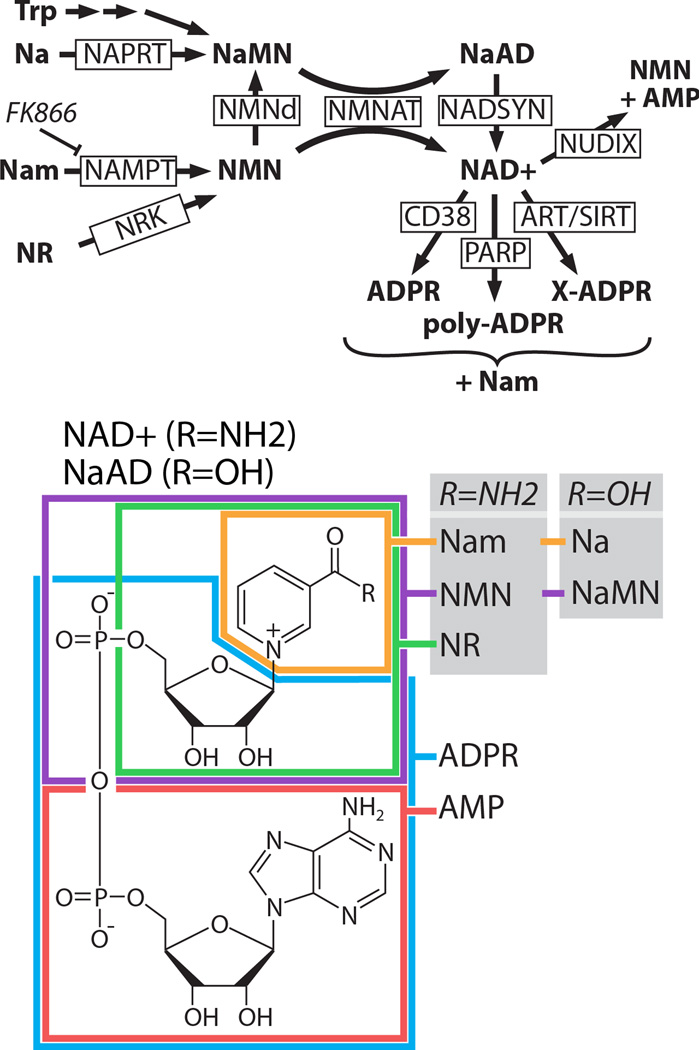
Pathways of NAD+ synthesis and breakdown. TOP: NAD+ is synthesized from nicotinamide (Nam), nicotinic acid (Na), nicotinamide riboside (NR), or tryptophan (Trp). All synthetic pathways require NMNAT. Nicotinic acid mononucleotide (NaMN) can be synthesized from nicotinamide mononucleotide (NMN) in some bacteria by the enzyme NMN deaminase (NMNd), which has no mammalian ortholog. NAD+ is broken down by multiple classes of enzymes including the glycohydrolase CD38 (Aksoy et al., 2006), poly-ADP-ribose polymerases (PARPs), NUDIX phosphohydrolases (McLennan, 2006), ADP ribosyltransferases (ARTs), and sirtuins (SIRTs). PARPs create polymers of ADPR that are usually attached to a protein substrate. ARTs transfer adenosine diphosphate ribose (ADPR) from NAD+ to an acceptor molecule (X) such as a protein. SIRTs transfer an O-acetyl group from a protein substrate to the ADPR moiety of NAD+ to yield O-acetyl-ADPR and Nam. NaMN=nicotinic acid mononucleotide; NaAD=nicotinic acid adenine dinucleotide; NMN=nicotinamide mononucleotide; NAD+=nicotinamide adenine dinucleotide; AMP=adenosine monophosphate; NAPRT=nicotinic acid phosphoribosyltransferase, NMNAT=nicotinamide mononucleotide adenyltransferase, NADSYN=NAD synthetase, NAMPT=nicotinamide phosphoribosyltransferase; NRK=nicotinamide riboside kinase, NUDIX=nucleoside diphosphate moiety linked X. BOTTOM: Structure of NAD+ with substrate moieties approximately outlined. For a more detailed overview of this pathway, please see these reviews: (Belenky et al., 2007; Chiarugi et al., 2012).
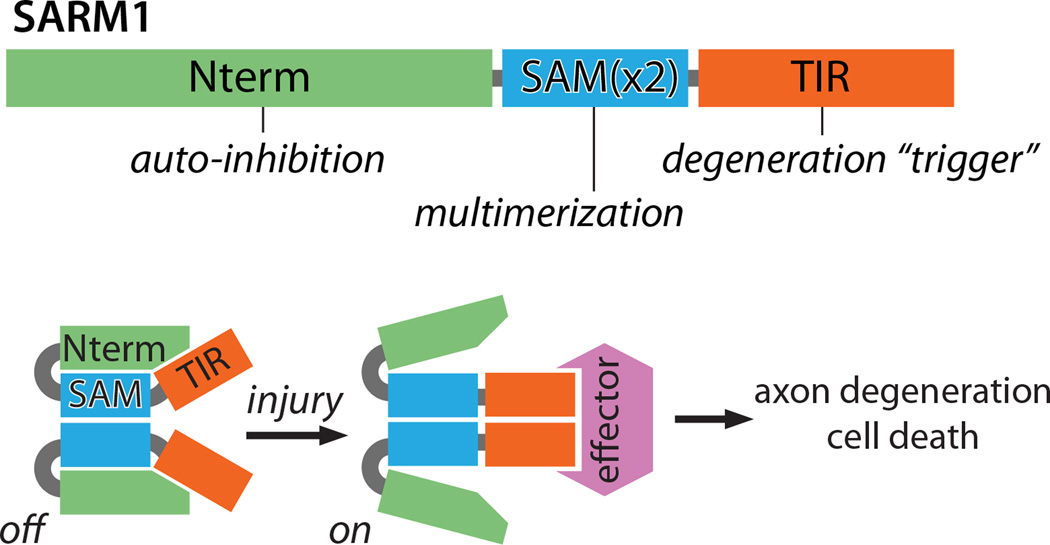
A working model of SARM1 auto-inhibition and activation upon injury. TOP: SARM1 is made up of three regions: 1) an auto-inhibitory N terminus (Nterm) comprised of multiple armadillo repeat motifs, 2) tandem SAM domains that mediate SARM1-SARM1 binding (SAMx2), and 3) a TIR domain that triggers axon degeneration upon multimerization. BOTTOM: SARM1 multimers are inactive (auto-inhibited) in uninjured axons. Injury leads to SARM1 activation, perhaps through release of inhibition, exposing TIR domain multimers that transmit a pro-destructive signal to unknown effector molecule(s).
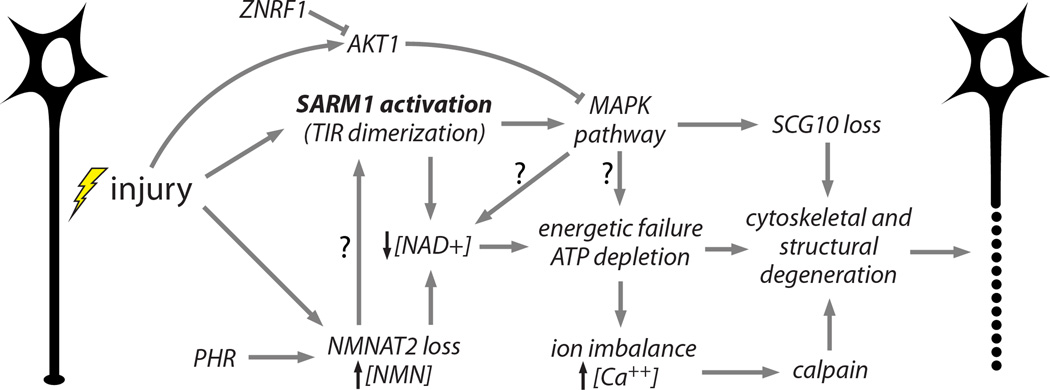
Working model of an integrated axon degeneration signaling cascade. Injury leads to SARM1 activation (Osterloh et al., 2012) and NMNAT2 depletion (Gilley and Coleman, 2010). PHR1 promotes NMNAT2 turnover, leading to faster depletion (Babetto et al., 2013; Xiong et al., 2012). Activated SARM1 promotes NAD+ depletion (Gerdts et al., 2015) and NMNAT2 loss prevents NAD+ synthesis and causes an increased NMN to ATP+ADP+AMP ratio, which may activate SARM1 (Gilley et al., 2015). NAD+ loss leads to glycolytic failure and ATP depletion. SARM1 also activates MAPK pathway signaling (Yang et al., 2015), which promotes SCG10 proteolysis (Shin et al., 2012a) and contributes to ATP depletion, perhaps via NAD+ depletion. MAPK activation is counteracted by injury-stimulated ATK1 activity (Yang et al., 2015), and AKT is in turn destabilized by ZNRF1 (Wakatsuki et al., 2011). Energetic failure promotes ionic imbalance including intraaxonal calcium accumulation, leading to calpain activation and proteolysis of intermediate filaments in the axonal cytoskeleton (Yang et al., 2013). Cumulative structural damage leads to irreversible fragmentation of the damaged axon (Wang et al., 2012). Arrows with questions marks (?) reflect postulated interactions.
Similar articles
-
SARM1 activation triggers axon degeneration locally via NAD⁺ destruction.Science. 2015 Apr 24;348(6233):453-7. doi: 10.1126/science.1258366. Epub 2015 Apr 23. Science. 2015. PMID: 25908823 Free PMC article.
-
Mitochondrial impairment activates the Wallerian pathway through depletion of NMNAT2 leading to SARM1-dependent axon degeneration.Neurobiol Dis. 2020 Feb;134:104678. doi: 10.1016/j.nbd.2019.104678. Epub 2019 Nov 15. Neurobiol Dis. 2020. PMID: 31740269 Free PMC article.
-
Protective effects of NAMPT or MAPK inhibitors and NaR on Wallerian degeneration of mammalian axons.Neurobiol Dis. 2022 Sep;171:105808. doi: 10.1016/j.nbd.2022.105808. Epub 2022 Jun 30. Neurobiol Dis. 2022. PMID: 35779777 Free PMC article.
-
The SARM1 axon degeneration pathway: control of the NAD+ metabolome regulates axon survival in health and disease.Curr Opin Neurobiol. 2020 Aug;63:59-66. doi: 10.1016/j.conb.2020.02.012. Epub 2020 Apr 17. Curr Opin Neurobiol. 2020. PMID: 32311648 Free PMC article. Review.
-
The chemical biology of NAD+ regulation in axon degeneration.Curr Opin Chem Biol. 2022 Aug;69:102176. doi: 10.1016/j.cbpa.2022.102176. Epub 2022 Jul 1. Curr Opin Chem Biol. 2022. PMID: 35780654 Free PMC article. Review.
Cited by
-
Multifaceted roles of SARM1 in axon degeneration and signaling.Front Cell Neurosci. 2022 Aug 25;16:958900. doi: 10.3389/fncel.2022.958900. eCollection 2022. Front Cell Neurosci. 2022. PMID: 36090788 Free PMC article. Review.
-
Wnd/DLK Is a Critical Target of FMRP Responsible for Neurodevelopmental and Behavior Defects in the Drosophila Model of Fragile X Syndrome.Cell Rep. 2019 Sep 3;28(10):2581-2593.e5. doi: 10.1016/j.celrep.2019.08.001. Cell Rep. 2019. PMID: 31484070 Free PMC article.
-
Degeneration of Injured Axons and Dendrites Requires Restraint of a Protective JNK Signaling Pathway by the Transmembrane Protein Raw.J Neurosci. 2019 Oct 23;39(43):8457-8470. doi: 10.1523/JNEUROSCI.0016-19.2019. Epub 2019 Sep 6. J Neurosci. 2019. PMID: 31492772 Free PMC article.
-
Cellular Compartmentation and the Redox/Nonredox Functions of NAD.Antioxid Redox Signal. 2019 Sep 20;31(9):623-642. doi: 10.1089/ars.2018.7722. Epub 2019 Mar 26. Antioxid Redox Signal. 2019. PMID: 30784294 Free PMC article.
-
Untangling the Tauopathy for Alzheimer's disease and parkinsonism.J Biomed Sci. 2018 Jul 10;25(1):54. doi: 10.1186/s12929-018-0457-x. J Biomed Sci. 2018. PMID: 29991349 Free PMC article. Review.
References
-
- Aksoy P, White Ta, Thompson M, Chini EN. Regulation of intracellular levels of NAD: A novel role for CD38. Biochem. Biophys. Res. Commun. 2006;345:1386–1392. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
