

Enzyme replacement therapy with idursulfase for mucopolysaccharidosis type II (Hunter syndrome)
- PMID: 26845288
- PMCID: PMC7173756
- DOI: 10.1002/14651858.CD008185.pub4
Enzyme replacement therapy with idursulfase for mucopolysaccharidosis type II (Hunter syndrome)
Abstract
Background: Mucopolysaccharidosis II, also known as Hunter syndrome, is a rare, X-linked disease caused by a deficiency of the lysosomal enzyme iduronate-2-sulfatase, which catalyses a step in the catabolism of glycosaminoglycans. The glycosaminoglycans accumulate within tissues affecting multiple organs and physiologic systems. The clinical manifestations include neurologic involvement, severe airways obstruction, skeletal deformities and cardiomyopathy. The disease has a variable age of onset and variable rate of progression. In those with severe disease, death usually occurs in the second decade of life, whereas those individuals with less severe disease may survive into adulthood. Enzyme replacement therapy with intravenous infusions of idursulfase has emerged as a new treatment for mucopolysaccharidosis type II. This is an update of a previously published version of this review.
Objectives: To evaluate the effectiveness and safety of enzyme replacement therapy with idursulfase compared to other interventions, placebo or no intervention, for treating mucopolysaccharidosis type II.
Search methods: We searched the Cochrane Cystic Fibrosis and Genetic Disorders Group's Trials Register (date of last search 23 November 2015).We also searched Embase, PubMed and the Literature Latino-Americana e do Caribe em Ciências da Saúde (LILACS) (date of last search 28 November 2015).
Selection criteria: Randomised and quasi-randomised controlled trials of enzyme replacement therapy with idursulfase compared to no intervention, placebo or other options (e.g. behavioral strategies, transplantation).
Data collection and analysis: Two authors independently screened the trials identified, appraised quality of papers and extracted data.
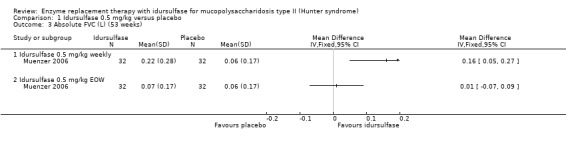
Main results: One study (96 male participants) met the inclusion criteria, although the primary outcome of this review - z score for height and weight, was not assessed in the study. This trial was considered to be of overall good quality. Following 53 weeks of treatment, participants in the weekly idursulfase 0.5 mg/kg group demonstrated a significant improvement rate compared with placebo for the primary outcome: distance walked in six minutes on the basis of the sum of ranks of change from baseline, mean difference 37.00 (95% confidence interval 6.52 to 67.48). The every-other-week idursulfase 0.5 mg/kg group also showed an improvement, which was not significant compared with placebo, mean difference 23.00 (95% confidence interval -4.49 to 50.49). After 53 weeks, there was no statistical significance difference in per cent predicted forced vital capacity between the three groups and absolute forced vital capacity was significantly increased from baseline in the weekly dosing group compared to placebo, mean difference 0.16 (95% confidence interval CI 0.05 to 0.27). No difference was observed between the every-other-week idursulfase 0.5 mg/kg group and placebo.In addition, liver and spleen volumes and urine glycosaminoglycan excretion were significantly reduced from baseline by both idursulfase dosing regimens. Idursulfase was generally well tolerated, but infusion reactions did occur. Idursulfase antibodies were detected in 31.7% of participants at the end of the study and they were related to a smaller reduction in urine glycosaminoglycan levels.
Authors' conclusions: The current evidence is limited. While the randomised clinical trial identified was considered to be of good quality, it failed to describe important outcomes. It has been demonstrated that enzyme replacement therapy with idursulfase is effective in relation to functional capacity (distance walked in six minutes and forced vital capacity), liver and spleen volumes and urine glycosaminoglycan excretion in people with mucopolysaccharidosis type II compared with placebo. There is no available evidence in the included study and in the literature on outcomes such as improvement in growth, sleep apnoea, cardiac function, quality of life and mortality. More studies are needed to obtain more information on the long-term effectiveness and safety of enzyme replacement therapy.
Conflict of interest statement
None known.
Figures










Update of
-
Enzyme replacement therapy with idursulfase for mucopolysaccharidosis type II (Hunter syndrome).Cochrane Database Syst Rev. 2014 Jan 8;(1):CD008185. doi: 10.1002/14651858.CD008185.pub3. Cochrane Database Syst Rev. 2014. Update in: Cochrane Database Syst Rev. 2016 Feb 05;2:CD008185. doi: 10.1002/14651858.CD008185.pub4. PMID: 24399699 Updated.
References
References to studies included in this review
Muenzer 2006 {published data only}
-
- Beck M, Wraith E, Muenzer J, Giugliani R, Harmatz P, Eng CM, et al. Long‐term weekly dosing of idursulfase in the treatment of mucopolysaccharidosis II (MPS II, Hunter syndrome) [abstract]. Journal of Inherited Metabolic Disease 2007;30(Suppl 1):116.
-
- Muenzer J, Beck M, Eng CM, Giugliani R, Harmatz P, Martin R, et al. Long‐term, open‐labeled extension study of idursulfase in the treatment of Hunter syndrome. Genetics in Medicine 2011;13(2):95‐101. - PubMed
-
- Muenzer J, Wraith E, Beck M, Giugliani R, Harmatz P, Eng CM, et al. Clinical benefit of enzyme replacement therapy (ERT) in mucopolysaccharidosis II (MPS II, Hunter syndrome) [abstract]. Journal of Inherited Metabolic Disease 2006;29(Suppl 1):28.
-
- Muenzer J, Wraith JE, Beck M, Giugliani R, Harmatz P, Eng CM, et al. A phase II/III clinical study of enzyme replacement therapy with idursulfase in mucopolysaccharidosis II (Hunter syndrome). Genetics in Medicine 2006;8(8):465‐73. - PubMed
References to studies excluded from this review
Gutiérrez‐Solana 2007 {published data only}
-
- Gutiérrez‐Solana LG. Clinical study of enzyme replacement therapy with idursulfase. Revista de Neurologia 2007;44(Suppl 1):S7‐S11. - PubMed
Muenzer 2007 {published data only}
-
- Muenzer J, Calikoglu MG, Conway AM, Kimura A. The long‐term experience of enzyme replacement therapy for Hunter syndrome (MPS II) [abstract]. Journal of Inherited Metabolic Disorder 2005;28(Suppl 1):188.
-
- Muenzer J, Gucsavas‐Calikoglu M, McCandless SE, Schuetz TJ, Kimura A. A phase I/II clinical trial of enzyme replacement therapy in mucopolysaccharidosis II (Hunter syndrome). Molecular Genetics and Metabolism. 2007;90(3):329‐37. - PubMed
-
- Muenzer J, Towle D, Calikoglu M, McCandless S. The 12‐month experience of enzyme replacement for mucopolysaccharidosis II (Hunter syndrome) [abstract]. Journal of Inherited Metabolic Disease 2003;26(Suppl 2):144.
Muenzer 2016 {published data only}
-
- Muenzer J, Hendriksz CJ, Fan Z, Vijayaraghavan S, Perry V4, Santra S, et al. A phase I/II study of intrathecal idursulfase‐IT in children with severe mucopolysaccharidosis II. Genetics in Medicine 2016;18(1):73‐81. - PubMed
Sohn 2013 {published data only}
-
- Sohn YB, Cho SY, Park SW, Kim SJ, Ko AR, Kwon EK, et al. Phase I/II clinical trial of enzyme replacement therapy with idursulfase beta in patients with mucopolysaccharidosis II (Hunter Syndrome) [abstract]. Journal of Inherited Metabolic Disease 2013;36 Suppl 2:S260, Abstract no: P‐565. [CENTRAL: 977628; CRS: 5500125000000518]
Sohn 2015 {published data only}
-
- Sohn YB, Cho SY, Lee J, Kwun Y, Huh R, Jin DK. Safety and efficacy of enzyme replacement therapy with idursulfase beta in children aged younger than 6 years with Hunter syndrome. Molecular Genetics and Metabolism 2015;114(2):156‐60. - PubMed
Tylki‐Szymanska 2008 {published data only}
-
- Tylki‐Szymanska A, Giugliani R, Hwu WL. A clinical trial of idursulfase in Hunter syndrome patients 5 years old and younger [abstract]. Journal of Inherited Metabolic Disease 2008;31(Suppl 1):130.
References to ongoing studies
NCT02055118 {published data only}
-
- NCT02055118. A controlled, randomized, two‐arm, open‐label, assessor‐blinded, multicenter study of intrathecal idursulfase‐IT administered in conjunction with elaprase® in pediatric patients with Hunter syndrome and early cognitive impairment. https://clinicaltrials.gov/ct2/show/NCT02055118 (accessed 20 Jan 2016).
Additional references
Alcade‐Martín 2010
-
- Alcalde‐Martín C, Muro‐Tudelilla JM, Cancho‐Candela R, Gutiérrez‐Solana LG, Pintos‐Morell G, Martí‐Herrero M, et al. First experience of enzyme replacement therapy with idursulfase in Spanish patients with Hunter syndrome under 5 years of age: Case observations from the Hunter Outcome Survey (HOS). European Journal of Medical Genetics 2010;53(6):371‐7. - PubMed
ATS 2002
-
- American Thoracic Society. ATS statement: guidelines for the six‐minute walk test. American Journal of Respiratory and Critical Care Medicine 2002;166(1):111‐7. - PubMed
de Jong 1992
-
- Jong JG, Wevers RA, Liebrand‐van Sambeek R. Measuring urinary glycosaminoglycans in the presence of protein: an improved screening procedure for mucopolysaccharidoses based on dimethylmethylene blue. Clinical Chemistry 1992;38(6):803‐7. - PubMed
Elbourne 2002
-
- Elbourne DR, Altman DG, Higgins JP, Curtin F, Worthington HV, Vail A. Meta‐analyses involving cross‐over trials: methodological issues. International Journal of Epidemiology 2002;31(1):140‐9. [PUBMED: 11914310] - PubMed
Higgins 2003
Higgins 2011a
-
- Higgins JPT, Altman DG. Chapter 8: Assessing risk of bias in included studies. In: Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1 [updated March 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
Higgins 2011b
-
- Higgins JPT, Deeks JJ, Altman DG. Chpater 16: Special topics in statistics. Intention to treat issues. In: Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1 [updated March 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
Jones 2008
-
- Jones SA, Almássy Z, Beck M, Burt K, Clarke JT, Giugliani R, et al. Mortality and cause of death in mucopolysaccharidosis type II‐a historical review based on data from the Hunter Outcome Survey (HOS). Journal of Inherited Metabolic Disease 2008;32(4):534‐43. - PubMed
Jones 2013
-
- Jones SA, Parini R, Harmatz P, Giugliani R, Fang J, Mendelsohn NJ, et al. The effect of idursulfase on growth in patients with Hunter syndrome: data from the Hunter Outcome Survey (HOS). Molecular Genetics and Metabolism 2013;109(1):41‐8. - PubMed
Kresse 1982
-
- Kresse H, Figura K, Klein U, Glossl J, Paschke E, Pohlmann R. Enzymic diagnosis of the genetic mucopolysaccharide storage disorders. Methods in Enzymology 1982;83:559‐72. - PubMed
Martin 2008
-
- Martin R, Beck M, Eng C, Giugliani R, Harmatz P, Munoz V, et al. Recognition and Diagnosis of Mucopolysaccharidosis II (Hunter Syndrome). Pediatrics 2008;121(2):e377‐86. - PubMed
Meikle 1999
-
- Meikle PJ, Hopwood JJ, Clague AE, Carey WF. Prevalence of lysosomal storage disorders. JAMA 1999;281(3):249‐54. - PubMed
Muenzer 2011
-
- Muenzer J, Beck M, Eng CM, Giugliani R, Harmatz P, Martin R, et al. Long‐term, open‐labeled extension study of idursulfase in the treatment of Hunter syndrome. Genetics in Medicine 2011;13(2):95‐101. - PubMed
National Horizon Scanning Centre 2005
-
- National Horizon Scanning Centre. New and Emerging Technology Briefing: Iduronate‐2‐sulfatase for Hunter Syndrome. The University of Birmingham 2005.
Neufeld 2001
-
- Neufeld EF, Muenzer J. The mucopolysaccharidoses. The Metabolic and Molecular Bases of Inherited Disease. New York, NY: McGraw‐Hill, 2001:3421‐52.
Schulze‐Frenking 2011
Tuschl 2005
-
- Tuschl K, Gal A, Paschke E, Kircher S, Bodamer OA. Mucopolysaccharidosis type II in females: case report and review of literature. Pediatric Neurology 2005;32(4):270‐2. - PubMed
Vellodi 2007
-
- Vellodi A, Wraith JE, Cleary M, Ramaswami U, Lavery C, Jessop E. Guidelines for the investigation and management of patients with mucopolysaccharidosis type II. www.specialisedservices.nhs.uk/library/23/Guidelines_for_Mucopolysacchar... (accessed September 2009).
Wraith 2008a
Wraith 2008b
-
- Wraith JE. Enzyme replacement therapy with idursulfase in patients with mucopolysaccharidosis type II. Acta Paediatrica. Supplementum 2008;97(497):76‐8. - PubMed
Young 1983
-
- Young ID, Harper PS. The natural history of the severe form of Hunter's syndrome: a study based on 52 cases. Developmental Medicine and Child Neurology 1983;25(4):481‐9. - PubMed
References to other published versions of this review
da Silva 2011
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous