

BS69/ZMYND11 C-Terminal Domains Bind and Inhibit EBNA2
- PMID: 26845565
- PMCID: PMC4742278
- DOI: 10.1371/journal.ppat.1005414
BS69/ZMYND11 C-Terminal Domains Bind and Inhibit EBNA2
Abstract
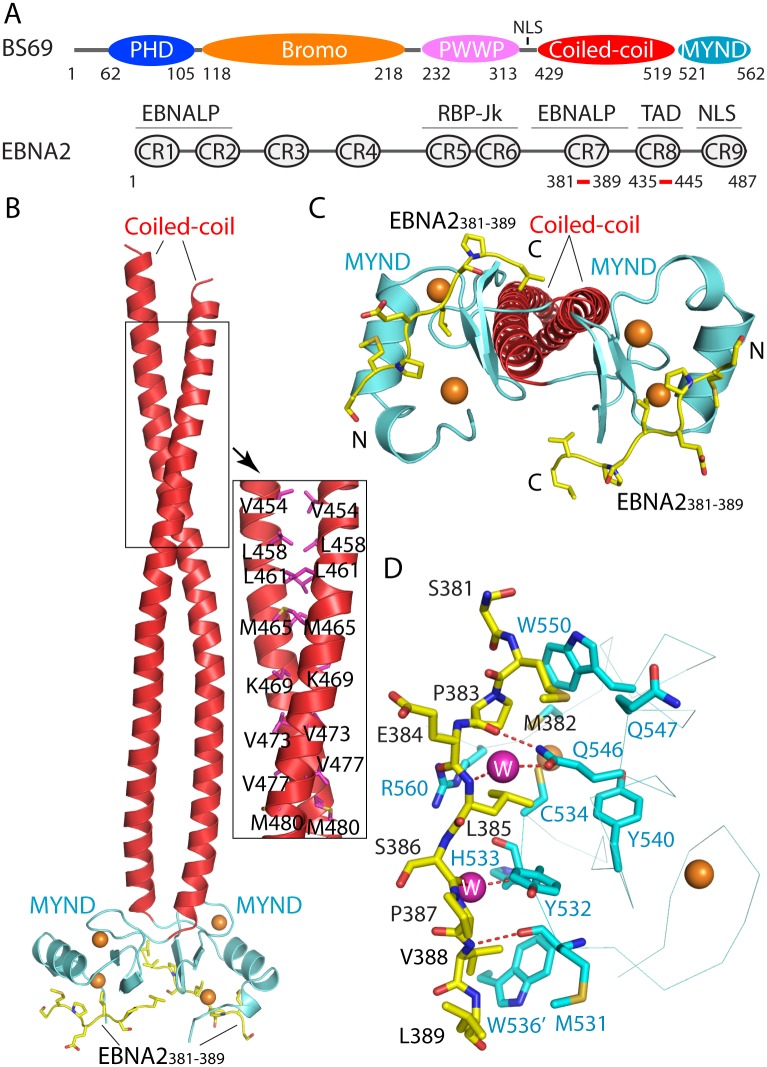
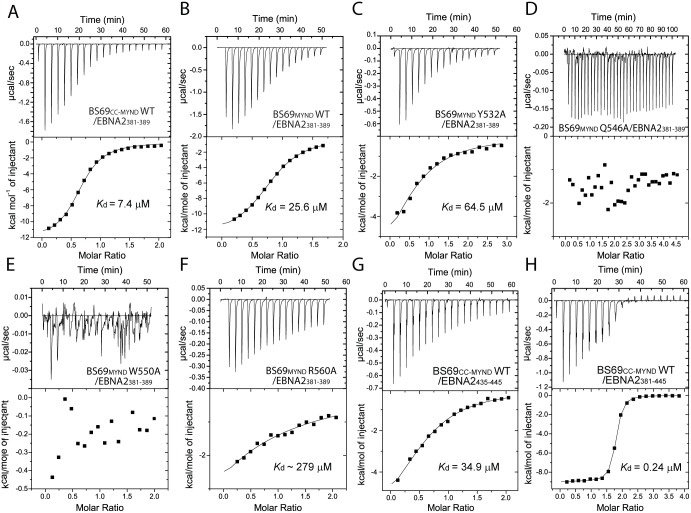
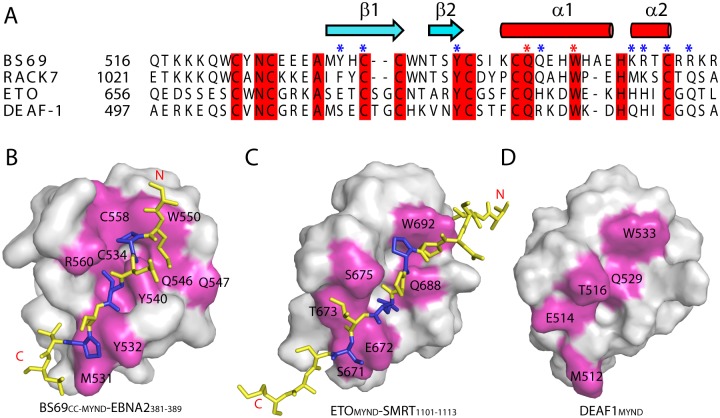
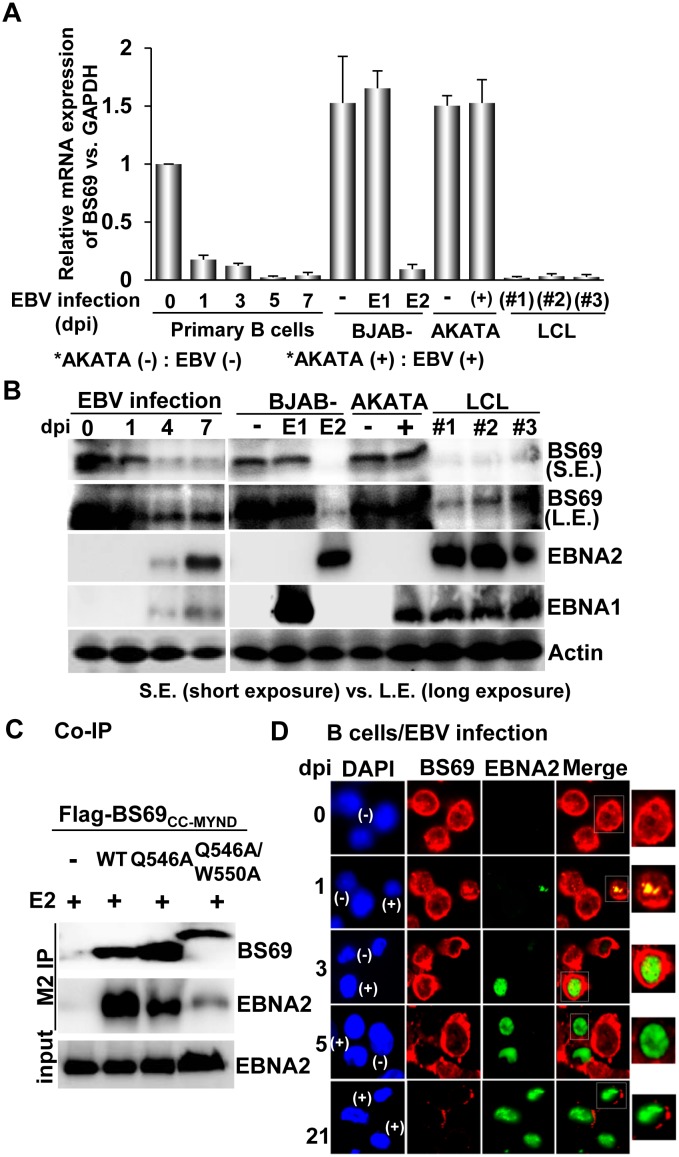
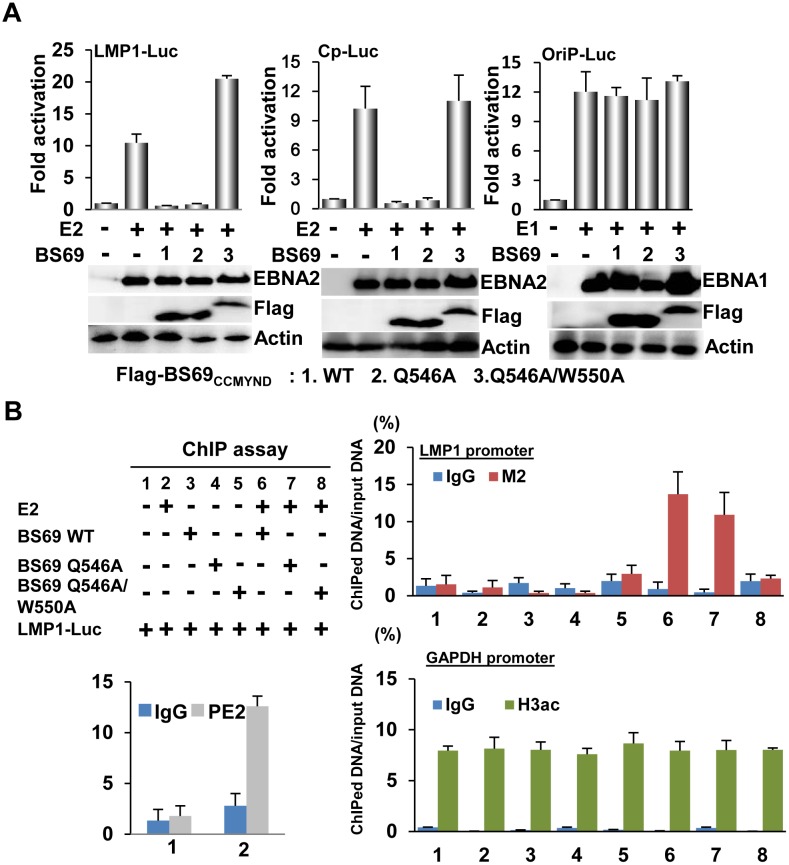
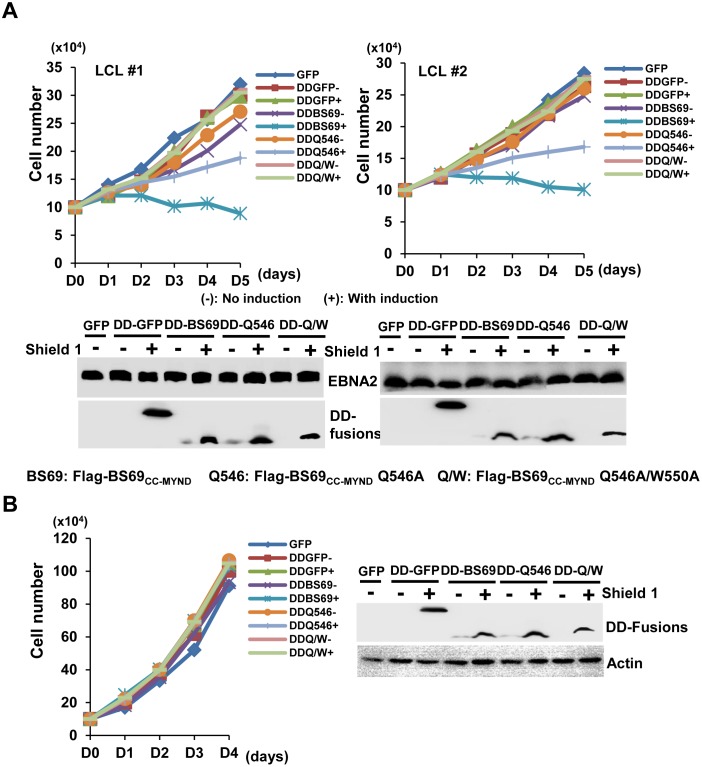
Epstein-Barr virus (EBV) nuclear antigen 2 (EBNA2) plays an important role in driving immortalization of EBV-infected B cells through regulating the expression of many viral and cellular genes. We report a structural study of the tumor suppressor BS69/ZMYND11 C-terminal region, comprised of tandem coiled-coil-MYND domains (BS69CC-MYND), in complex with an EBNA2 peptide containing a PXLXP motif. The coiled-coil domain of BS69 self-associates to bring two separate MYND domains in close proximity, thereby enhancing the BS69 MYND-EBNA2 interaction. ITC analysis of BS69CC-MYND with a C-terminal fragment of EBNA2 further suggests that the BS69CC-MYND homodimer synergistically binds to the two EBNA2 PXLXP motifs that are respectively located in the conserved regions CR7 and CR8. Furthermore, we showed that EBNA2 interacts with BS69 and down-regulates its expression at both mRNA and protein levels in EBV-infected B cells. Ectopic BS69CC-MYND is recruited to viral target promoters through interactions with EBNA2, inhibits EBNA2-mediated transcription activation, and impairs proliferation of lymphoblastoid cell lines (LCLs). Substitution of critical residues in the MYND domain impairs the BS69-EBNA2 interaction and abolishes the BS69 inhibition of the EBNA2-mediated transactivation and LCL proliferation. This study identifies the BS69 C-terminal domains as an inhibitor of EBNA2, which may have important implications in development of novel therapeutic strategies against EBV infection.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
Similar articles
-
Increased association between Epstein-Barr virus EBNA2 from type 2 strains and the transcriptional repressor BS69 restricts EBNA2 activity.PLoS Pathog. 2019 Jul 8;15(7):e1007458. doi: 10.1371/journal.ppat.1007458. eCollection 2019 Jul. PLoS Pathog. 2019. PMID: 31283782 Free PMC article.
-
The conserved Mynd domain of BS69 binds cellular and oncoviral proteins through a common PXLXP motif.J Biol Chem. 2002 Feb 15;277(7):4906-10. doi: 10.1074/jbc.M110078200. Epub 2001 Dec 3. J Biol Chem. 2002. PMID: 11733528
-
The nuclear chaperone nucleophosmin escorts an Epstein-Barr Virus nuclear antigen to establish transcriptional cascades for latent infection in human B cells.PLoS Pathog. 2012;8(12):e1003084. doi: 10.1371/journal.ppat.1003084. Epub 2012 Dec 13. PLoS Pathog. 2012. PMID: 23271972 Free PMC article.
-
Both Epstein-Barr viral nuclear antigen 2 (EBNA2) and activated Notch1 transactivate genes by interacting with the cellular protein RBP-J kappa.Immunobiology. 1997 Dec;198(1-3):299-306. doi: 10.1016/s0171-2985(97)80050-2. Immunobiology. 1997. PMID: 9442401 Review.
-
EBNA2 and Notch signalling in Epstein-Barr virus mediated immortalization of B lymphocytes.Semin Cancer Biol. 2001 Dec;11(6):423-34. doi: 10.1006/scbi.2001.0409. Semin Cancer Biol. 2001. PMID: 11669604 Review.
Cited by
-
ZMYND11-MBTD1 induces leukemogenesis through hijacking NuA4/TIP60 acetyltransferase complex and a PWWP-mediated chromatin association mechanism.Nat Commun. 2021 Feb 16;12(1):1045. doi: 10.1038/s41467-021-21357-3. Nat Commun. 2021. PMID: 33594072 Free PMC article.
-
Transcription factors operate across disease loci, with EBNA2 implicated in autoimmunity.Nat Genet. 2018 May;50(5):699-707. doi: 10.1038/s41588-018-0102-3. Epub 2018 Apr 16. Nat Genet. 2018. PMID: 29662164 Free PMC article.
-
Epstein-Barr Virus B Cell Growth Transformation: The Nuclear Events.Viruses. 2023 Mar 24;15(4):832. doi: 10.3390/v15040832. Viruses. 2023. PMID: 37112815 Free PMC article. Review.
-
Lysine Acetylation Goes Global: From Epigenetics to Metabolism and Therapeutics.Chem Rev. 2018 Feb 14;118(3):1216-1252. doi: 10.1021/acs.chemrev.7b00181. Epub 2018 Feb 6. Chem Rev. 2018. PMID: 29405707 Free PMC article. Review.
-
The Transcriptional Repressor BS69 is a Conserved Target of the E1A Proteins from Several Human Adenovirus Species.Viruses. 2018 Nov 22;10(12):662. doi: 10.3390/v10120662. Viruses. 2018. PMID: 30469473 Free PMC article.
References
-
- Epstein MA, Achong BG, & Barr YM (1964) Virus Particles in Cultured Lymphoblasts from Burkitt's Lymphoma. Lancet 1(7335):702–703. - PubMed
-
- Klein E, Kis LL, & Klein G (2007) Epstein-Barr virus infection in humans: from harmless to life endangering virus-lymphocyte interactions. Oncogene 26(9):1297–1305. - PubMed
-
- Rickinson AB & Kieff E (1996) Epstein-Barr virus Fields virology, eds Field BN, Knipe DM, & Howley PM (Lippincott-Raven Publishers, Philadelphia, Pa.), 3rd Ed, pp 2397–2446.
-
- Allday MJ, Crawford DH, & Griffin BE (1989) Epstein-Barr virus latent gene expression during the initiation of B cell immortalization. J Gen Virol 70 (Pt 7):1755–1764. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
