

c-Jun N-terminal kinase mediates mouse liver injury through a novel Sab (SH3BP5)-dependent pathway leading to inactivation of intramitochondrial Src
- PMID: 26845758
- PMCID: PMC4874901
- DOI: 10.1002/hep.28486
c-Jun N-terminal kinase mediates mouse liver injury through a novel Sab (SH3BP5)-dependent pathway leading to inactivation of intramitochondrial Src
Abstract
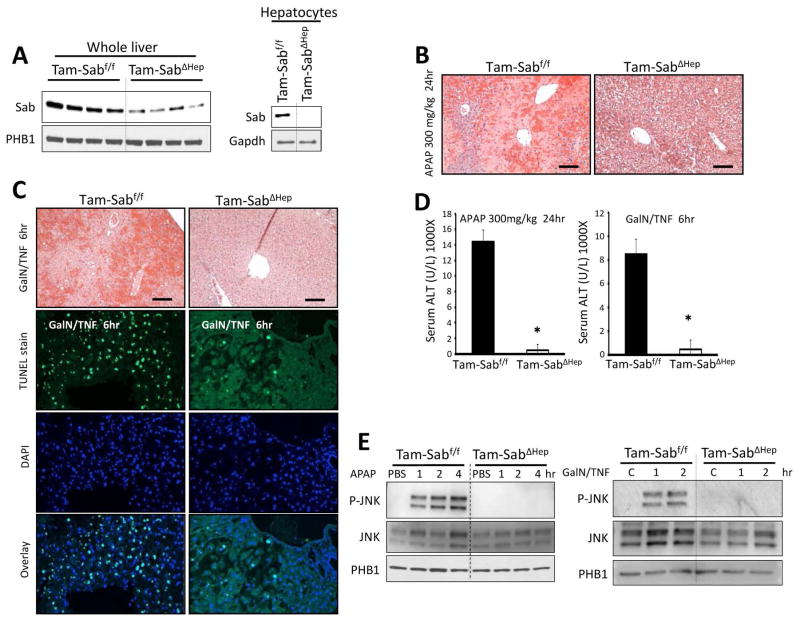
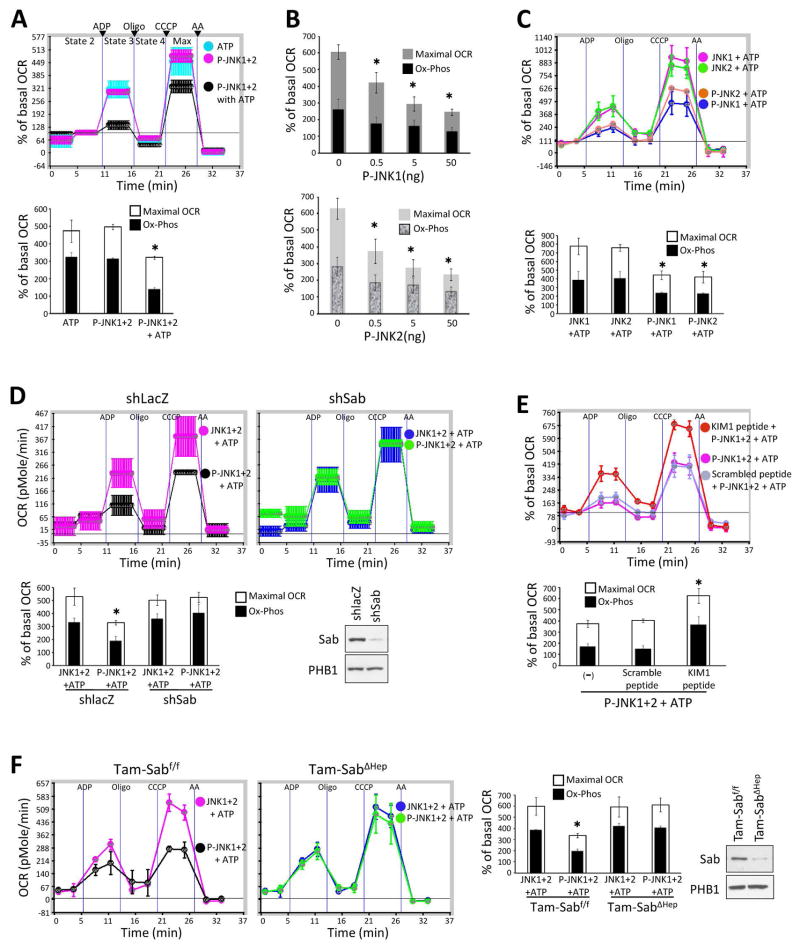
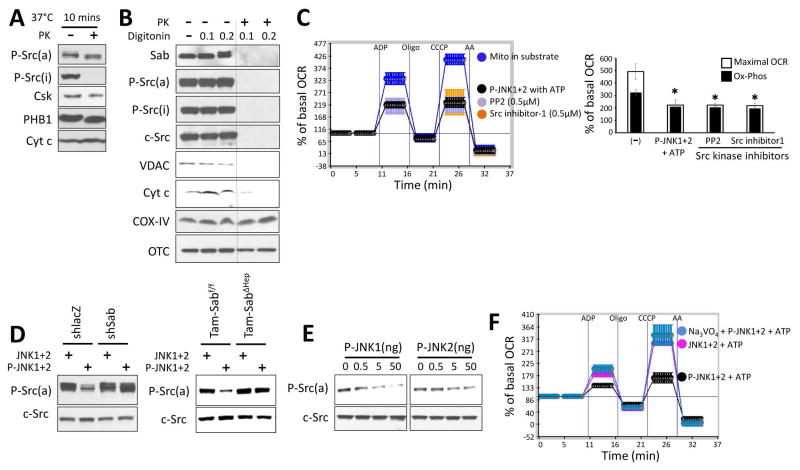
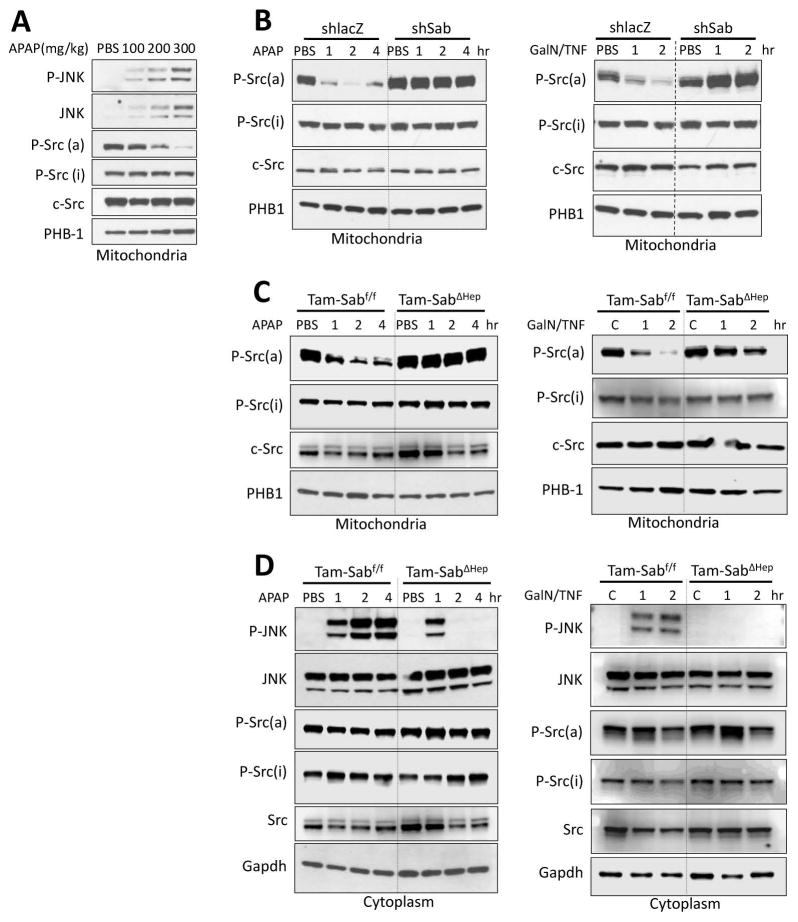
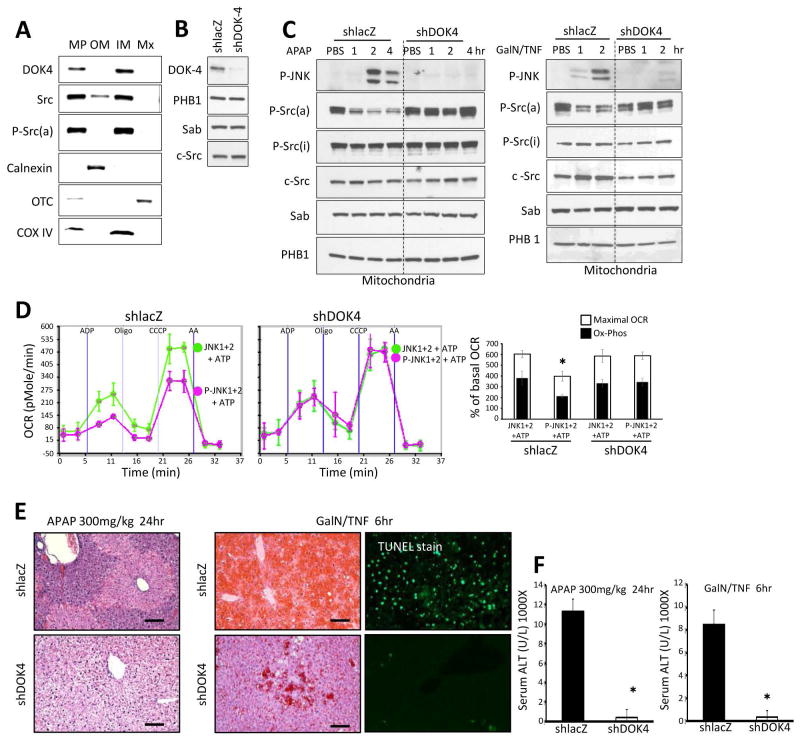
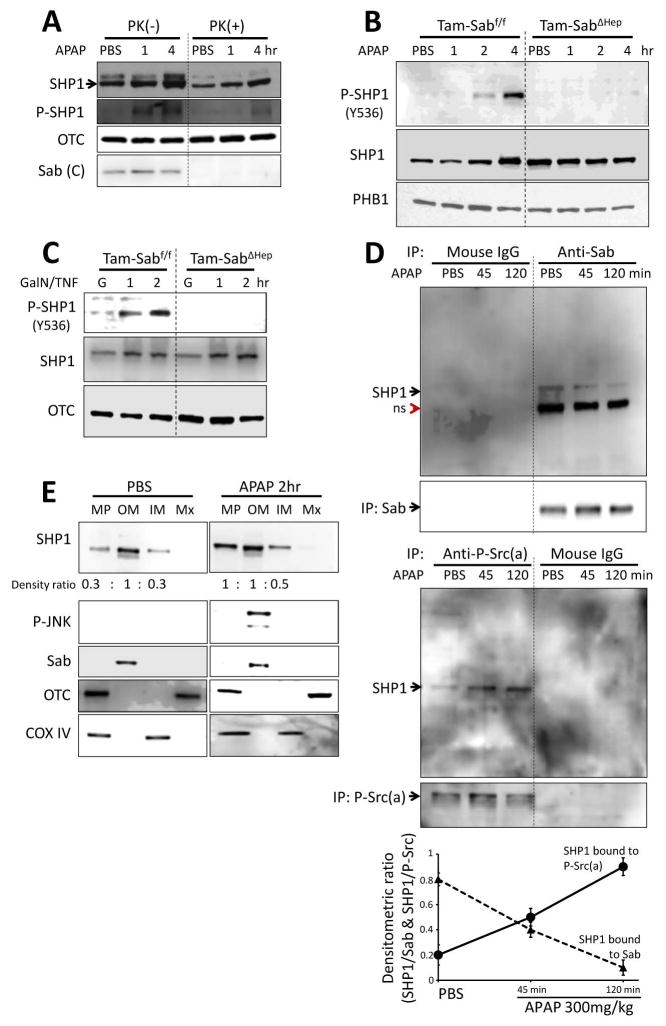
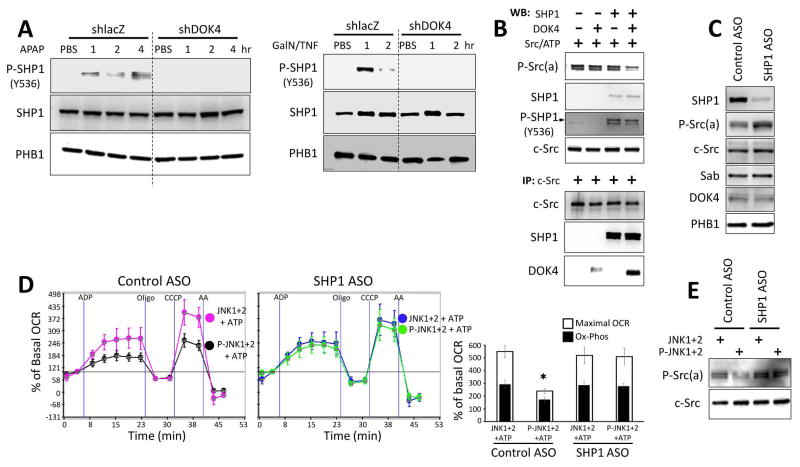
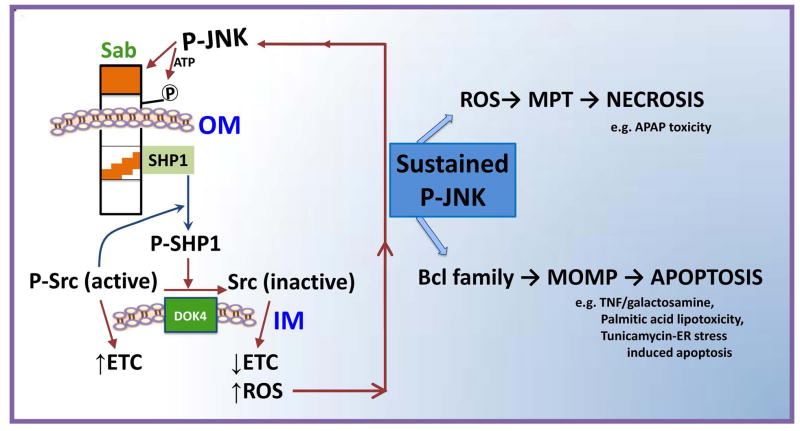
Sustained c-Jun N-terminal kinase (JNK) activation has been implicated in many models of cell death and tissue injury. Phosphorylated JNK (p-JNK) interacts with the mitochondrial outer membrane SH3 homology associated BTK binding protein (Sab, or SH3BP5). Using knockdown or liver-specific deletion of Sab, we aimed to elucidate the consequences of this interaction on mitochondrial function in isolated mitochondria and liver injury models in vivo. Respiration in isolated mitochondria was directly inhibited by p-JNK + adenosine triphosphate. Knockdown or liver-specific knockout of Sab abrogated this effect and markedly inhibited sustained JNK activation and liver injury from acetaminophen or tumor necrosis factor/galactosamine. We then elucidated an intramitochondrial pathway in which interaction of JNK and Sab on the outside of the mitochondria released protein tyrosine phosphatase, nonreceptor type 6 (SHP1, or PTPN6) from Sab in the inside of the mitochondrial outer membrane, leading to its activation and transfer to the inner membrane, where it dephosphorylates P-Y419Src (active), which required a platform protein, docking protein 4 (DOK4), on the inner membrane. Knockdown of mitochondrial DOK4 or SHP1 inhibited the inactivation of mitochondrial p-Src and the effect of p-JNK on mitochondria.
Conclusions: The binding to and phosphorylation of Sab by p-JNK on the outer mitochondrial membrane leads to SHP1-dependent and DOK4-dependent inactivation of p-Src on the inner membrane; inactivation of mitochondrial Src inhibits electron transport and increases reactive oxygen species release, which sustains JNK activation and promotes cell death and organ injury. (Hepatology 2016;63:1987-2003).
© 2016 by the American Association for the Study of Liver Diseases.
Conflict of interest statement
Conflict of interest statement: S.W, T.A.T, R.W.M.M and N.K have declared that no conflict of interest exists. M.A. is an employee and shareholder of Ionis Pharmaceuticals.
Figures
Similar articles
-
c-Jun N-terminal kinase (JNK)-dependent acute liver injury from acetaminophen or tumor necrosis factor (TNF) requires mitochondrial Sab protein expression in mice.J Biol Chem. 2011 Oct 7;286(40):35071-8. doi: 10.1074/jbc.M111.276089. Epub 2011 Aug 15. J Biol Chem. 2011. PMID: 21844199 Free PMC article.
-
Sab (Sh3bp5) dependence of JNK mediated inhibition of mitochondrial respiration in palmitic acid induced hepatocyte lipotoxicity.J Hepatol. 2015 Jun;62(6):1367-74. doi: 10.1016/j.jhep.2015.01.032. Epub 2015 Feb 7. J Hepatol. 2015. PMID: 25666017 Free PMC article.
-
The role of MAP2 kinases and p38 kinase in acute murine liver injury models.Cell Death Dis. 2017 Jun 29;8(6):e2903. doi: 10.1038/cddis.2017.295. Cell Death Dis. 2017. PMID: 28661486 Free PMC article.
-
The Regulation of JNK Signaling Pathways in Cell Death through the Interplay with Mitochondrial SAB and Upstream Post-Translational Effects.Int J Mol Sci. 2018 Nov 20;19(11):3657. doi: 10.3390/ijms19113657. Int J Mol Sci. 2018. PMID: 30463289 Free PMC article. Review.
-
Mitochondrial P-JNK target, SAB (SH3BP5), in regulation of cell death.Front Cell Dev Biol. 2024 Mar 15;12:1359152. doi: 10.3389/fcell.2024.1359152. eCollection 2024. Front Cell Dev Biol. 2024. PMID: 38559813 Free PMC article. Review.
Cited by
-
Suppressors of Superoxide-H2O2 Production at Site IQ of Mitochondrial Complex I Protect against Stem Cell Hyperplasia and Ischemia-Reperfusion Injury.Cell Metab. 2016 Oct 11;24(4):582-592. doi: 10.1016/j.cmet.2016.08.012. Epub 2016 Sep 22. Cell Metab. 2016. PMID: 27667666 Free PMC article.
-
Adipose-Derived Mesenchymal Stem Cells Inhibit JNK-Mediated Mitochondrial Retrograde Pathway to Alleviate Acetaminophen-Induced Liver Injury.Antioxidants (Basel). 2023 Jan 9;12(1):158. doi: 10.3390/antiox12010158. Antioxidants (Basel). 2023. PMID: 36671020 Free PMC article.
-
Peroxiredoxin 6 mediates acetaminophen-induced hepatocyte death through JNK activation.Redox Biol. 2020 May;32:101496. doi: 10.1016/j.redox.2020.101496. Epub 2020 Mar 8. Redox Biol. 2020. PMID: 32171727 Free PMC article.
-
Mechanisms of acetaminophen hepatotoxicity and their translation to the human pathophysiology.J Clin Transl Res. 2017 Feb;3(Suppl 1):157-169. doi: 10.18053/jctres.03.2017S1.002. Epub 2017 Feb 12. J Clin Transl Res. 2017. PMID: 28670625 Free PMC article.
-
Gallic Acid Alleviates Acetaminophen-Induced Acute Liver Injury by Regulating Inflammatory and Oxidative Stress Signaling Proteins.Antioxidants (Basel). 2025 Jul 14;14(7):860. doi: 10.3390/antiox14070860. Antioxidants (Basel). 2025. PMID: 40722964 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
