

Characterization of chromatin accessibility with a transposome hypersensitive sites sequencing (THS-seq) assay
- PMID: 26846207
- PMCID: PMC4743176
- DOI: 10.1186/s13059-016-0882-7
Characterization of chromatin accessibility with a transposome hypersensitive sites sequencing (THS-seq) assay
Abstract
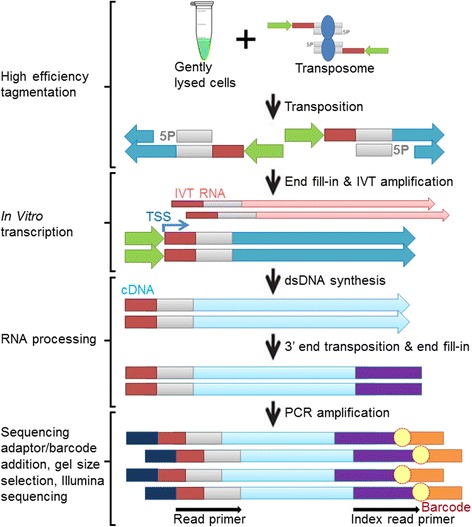
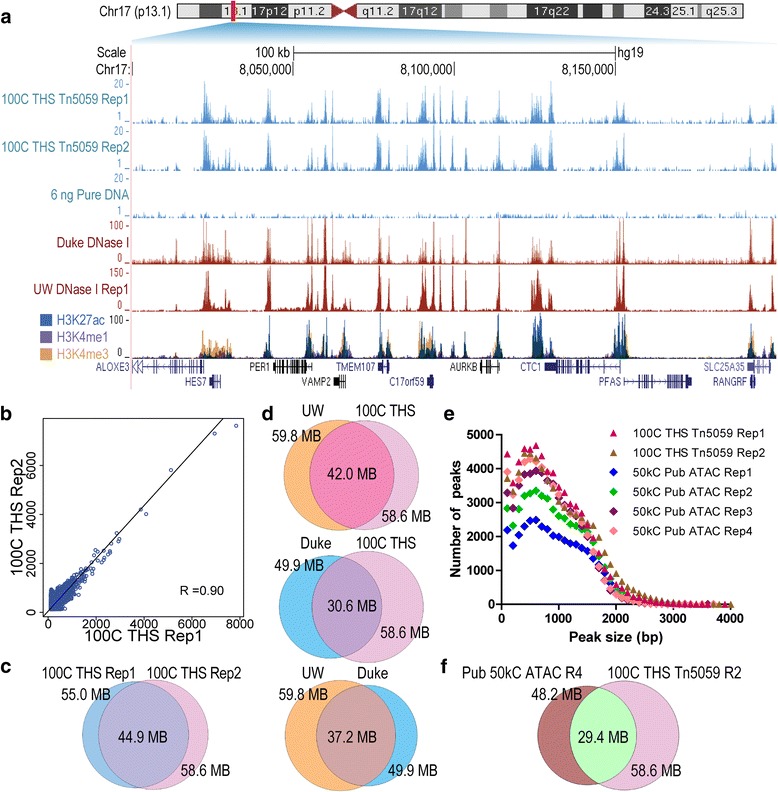
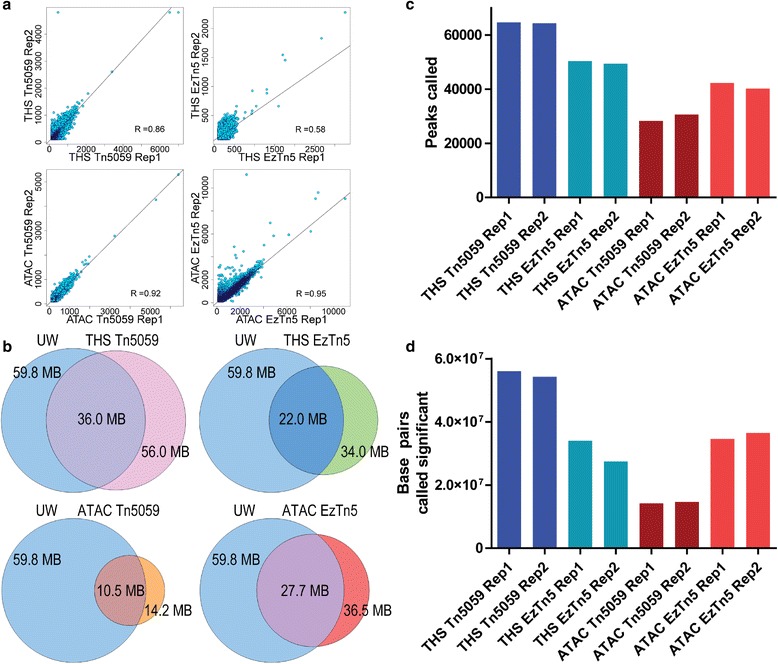
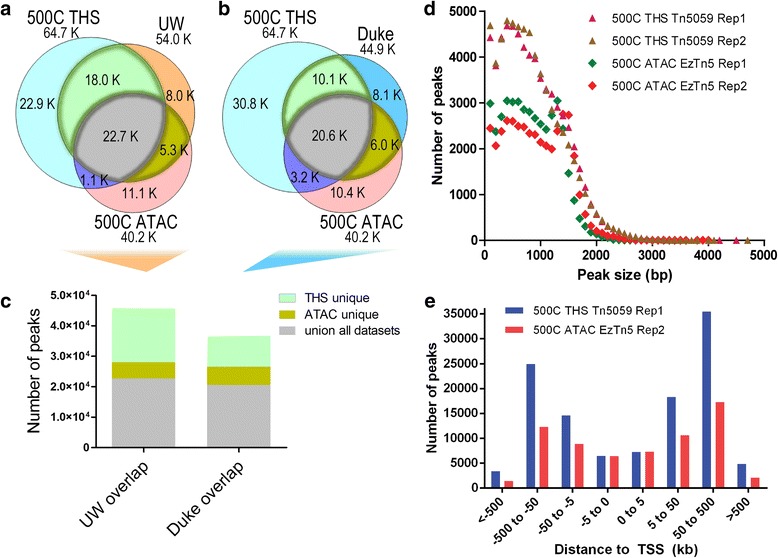
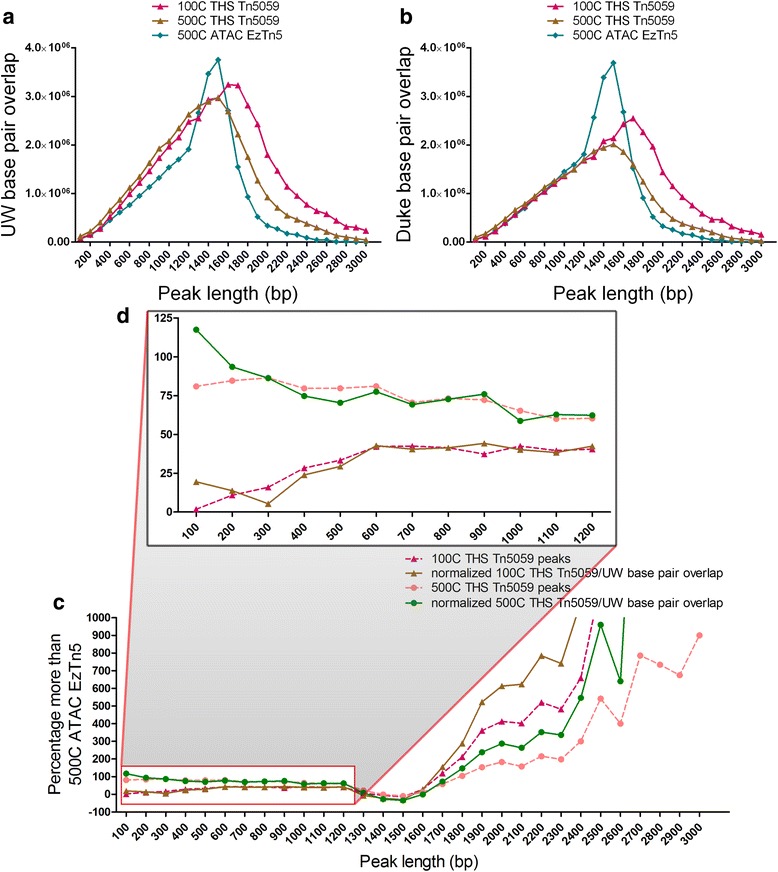
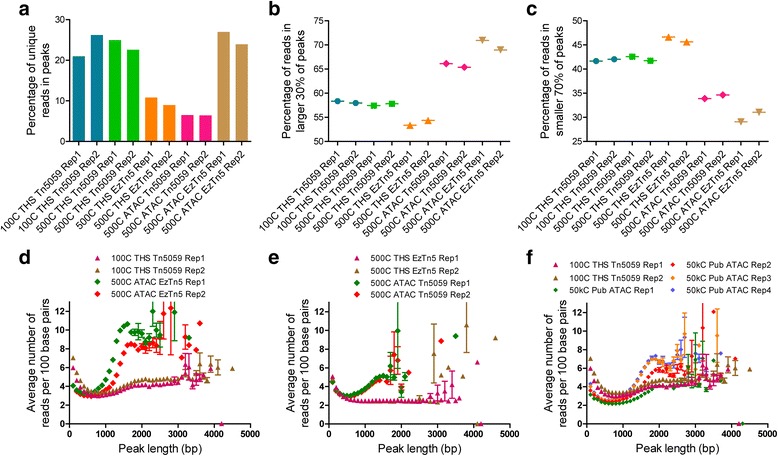
Chromatin accessibility captures in vivo protein-chromosome binding status, and is considered an informative proxy for protein-DNA interactions. DNase I and Tn5 transposase assays require thousands to millions of fresh cells for comprehensive chromatin mapping. Applying Tn5 tagmentation to hundreds of cells results in sparse chromatin maps. We present a transposome hypersensitive sites sequencing assay for highly sensitive characterization of chromatin accessibility. Linear amplification of accessible DNA ends with in vitro transcription, coupled with an engineered Tn5 super-mutant, demonstrates improved sensitivity on limited input materials, and accessibility of small regions near distal enhancers, compared with ATAC-seq.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
