

Molecular Pathological Classification of Neurodegenerative Diseases: Turning towards Precision Medicine
- PMID: 26848654
- PMCID: PMC4783923
- DOI: 10.3390/ijms17020189
Molecular Pathological Classification of Neurodegenerative Diseases: Turning towards Precision Medicine
Abstract
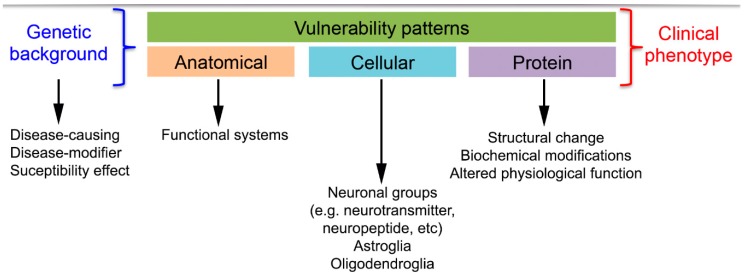
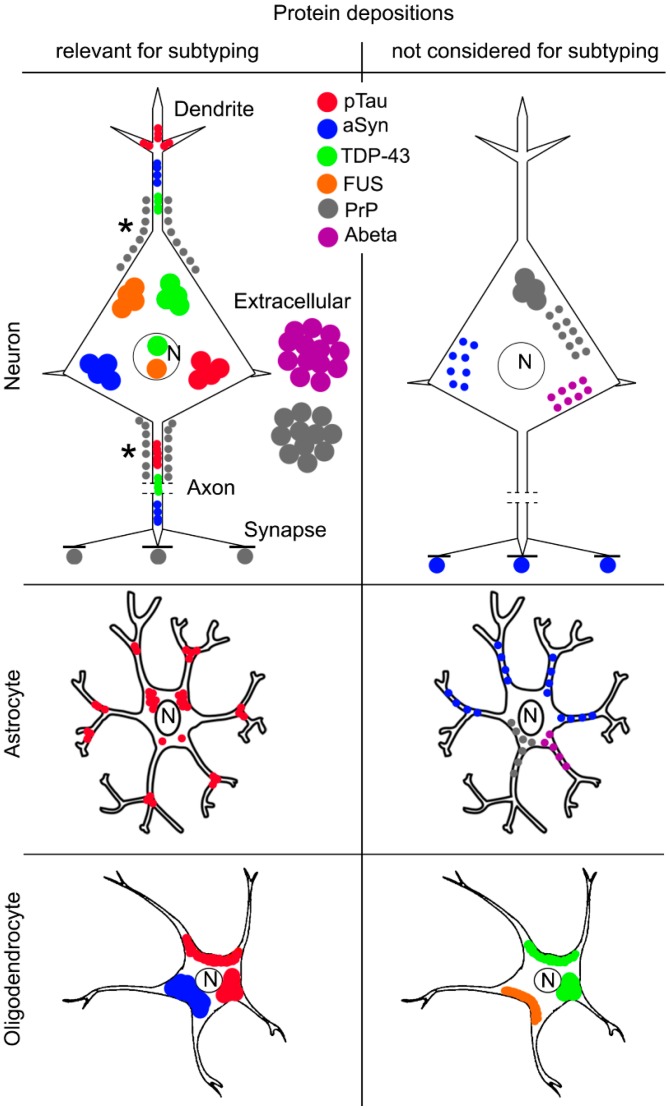
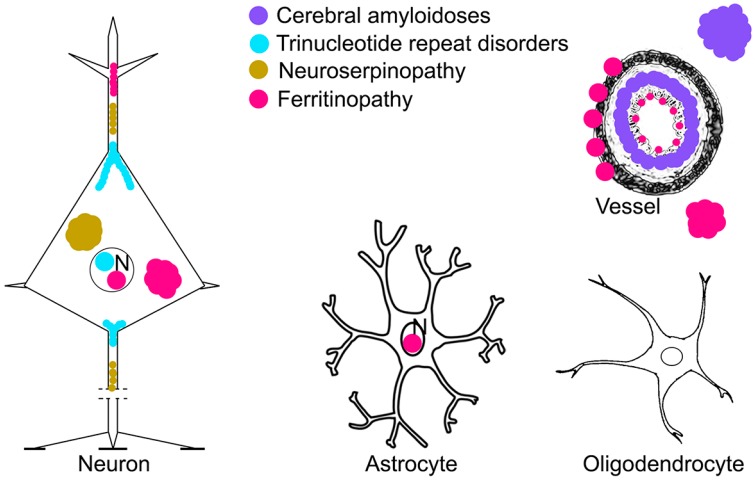
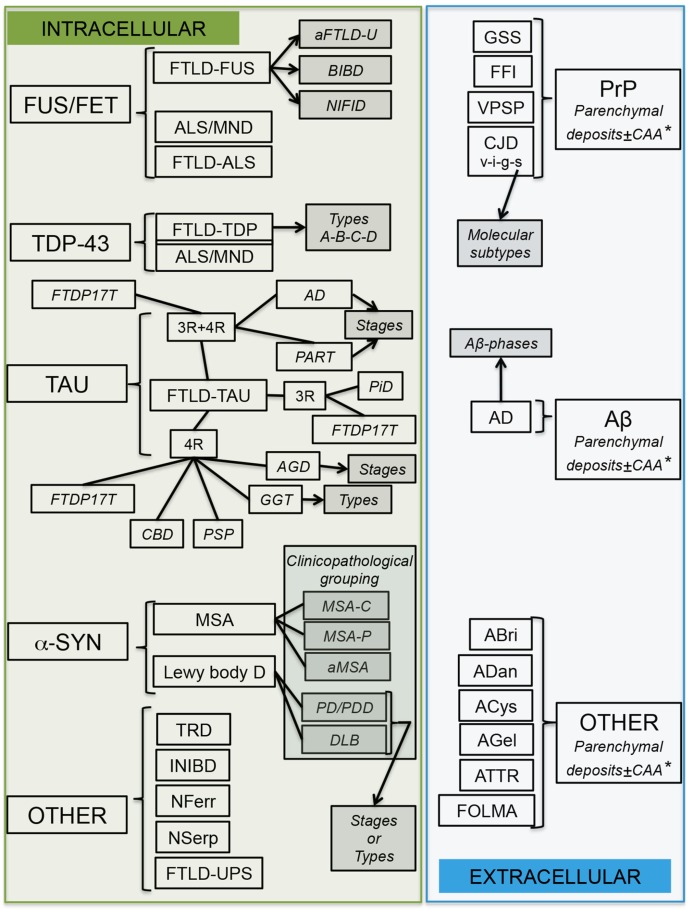
Neurodegenerative diseases (NDDs) are characterized by selective dysfunction and loss of neurons associated with pathologically altered proteins that deposit in the human brain but also in peripheral organs. These proteins and their biochemical modifications can be potentially targeted for therapy or used as biomarkers. Despite a plethora of modifications demonstrated for different neurodegeneration-related proteins, such as amyloid-β, prion protein, tau, α-synuclein, TAR DNA-binding protein 43 (TDP-43), or fused in sarcoma protein (FUS), molecular classification of NDDs relies on detailed morphological evaluation of protein deposits, their distribution in the brain, and their correlation to clinical symptoms together with specific genetic alterations. A further facet of the neuropathology-based classification is the fact that many protein deposits show a hierarchical involvement of brain regions. This has been shown for Alzheimer and Parkinson disease and some forms of tauopathies and TDP-43 proteinopathies. The present paper aims to summarize current molecular classification of NDDs, focusing on the most relevant biochemical and morphological aspects. Since the combination of proteinopathies is frequent, definition of novel clusters of patients with NDDs needs to be considered in the era of precision medicine. Optimally, neuropathological categorizing of NDDs should be translated into in vivo detectable biomarkers to support better prediction of prognosis and stratification of patients for therapy trials.
Keywords: biomarker; classification; molecular pathology; neurodegenerative disease; proteinopathy.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
