

The HTT CAG-Expansion Mutation Determines Age at Death but Not Disease Duration in Huntington Disease
- PMID: 26849111
- PMCID: PMC4746370
- DOI: 10.1016/j.ajhg.2015.12.018
The HTT CAG-Expansion Mutation Determines Age at Death but Not Disease Duration in Huntington Disease
Abstract
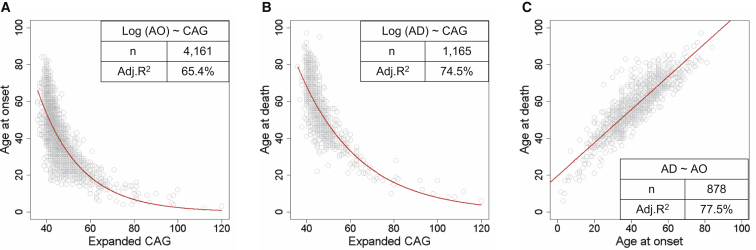
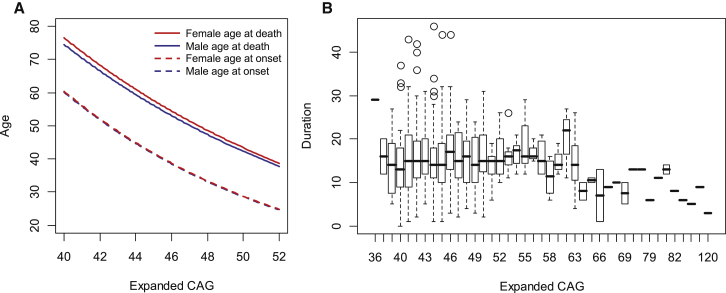
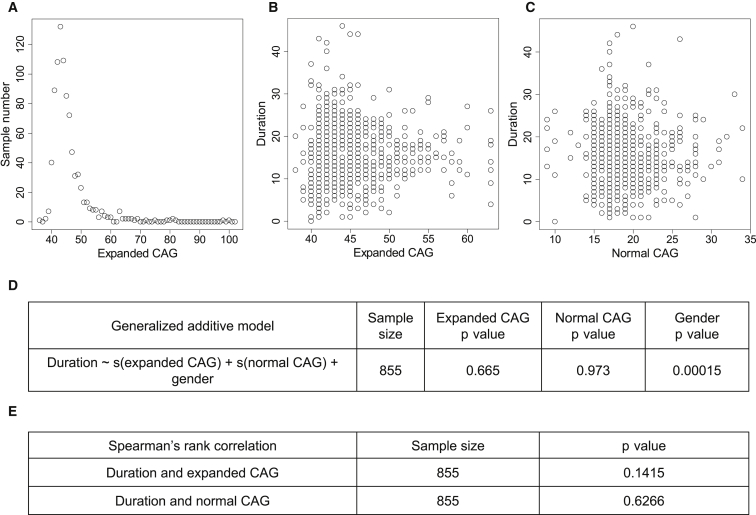
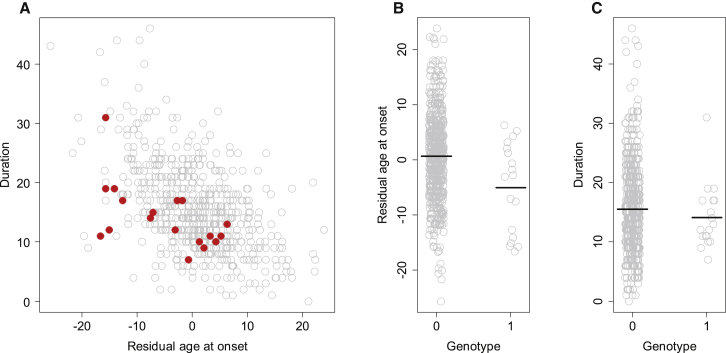
Huntington disease (HD) is caused by an expanded HTT CAG repeat that leads in a length-dependent, completely dominant manner to onset of a characteristic movement disorder. HD also displays early mortality, so we tested whether the expanded CAG repeat exerts a dominant influence on age at death and on the duration of clinical disease. We found that, as with clinical onset, HD age at death is determined by expanded CAG-repeat length and has no contribution from the normal CAG allele. Surprisingly, disease duration is independent of the mutation's length. It is also unaffected by a strong genetic modifier of HD motor onset. These findings suggest two parsimonious alternatives. (1) HD pathogenesis is driven by mutant huntingtin, but before or near motor onset, sufficient CAG-driven damage occurs to permit CAG-independent processes and then lead to eventual death. In this scenario, some pathological changes and their clinical correlates could still worsen in a CAG-driven manner after disease onset, but these CAG-related progressive changes do not themselves determine duration. Alternatively, (2) HD pathogenesis is driven by mutant huntingtin acting in a CAG-dependent manner with different time courses in multiple cell types, and the cellular targets that lead to motor onset and death are different and independent. In this scenario, processes driven by HTT CAG length lead directly to death but not via the striatal pathology associated with motor manifestations. Each scenario has important ramifications for the design and testing of potential therapeutics, especially those aimed at preventing or delaying characteristic motor manifestations.
Copyright © 2016 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures

References
-
- Huntington G. On chorea. Med. Surg. Rep. 1872;26:320–321.
-
- The Huntington’s Disease Collaborative Research Group A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell. 1993;72:971–983. - PubMed
-
- Cattaneo E., Zuccato C., Tartari M. Normal huntingtin function: an alternative approach to Huntington’s disease. Nat. Rev. Neurosci. 2005;6:919–930. - PubMed
-
- Bates G.P. History of genetic disease: the molecular genetics of Huntington disease - a history. Nat. Rev. Genet. 2005;6:766–773. - PubMed
-
- Gusella J., MacDonald M. No post-genetics era in human disease research. Nat. Rev. Genet. 2002;3:72–79. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
