

Gene Transfer of Brain-derived Neurotrophic Factor (BDNF) Prevents Neurodegeneration Triggered by FXN Deficiency
- PMID: 26849417
- PMCID: PMC4881769
- DOI: 10.1038/mt.2016.32
Gene Transfer of Brain-derived Neurotrophic Factor (BDNF) Prevents Neurodegeneration Triggered by FXN Deficiency
Abstract
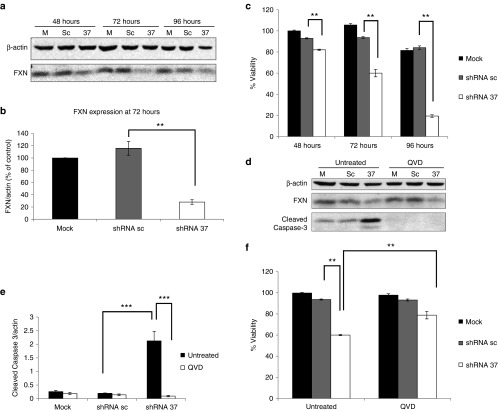
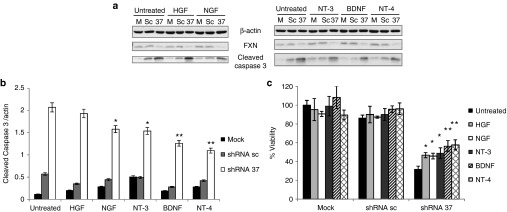
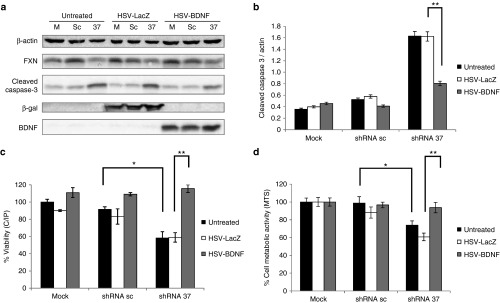
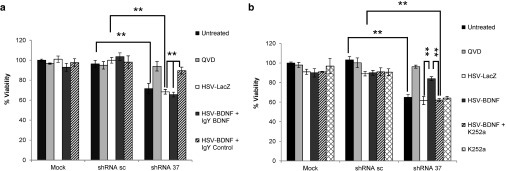
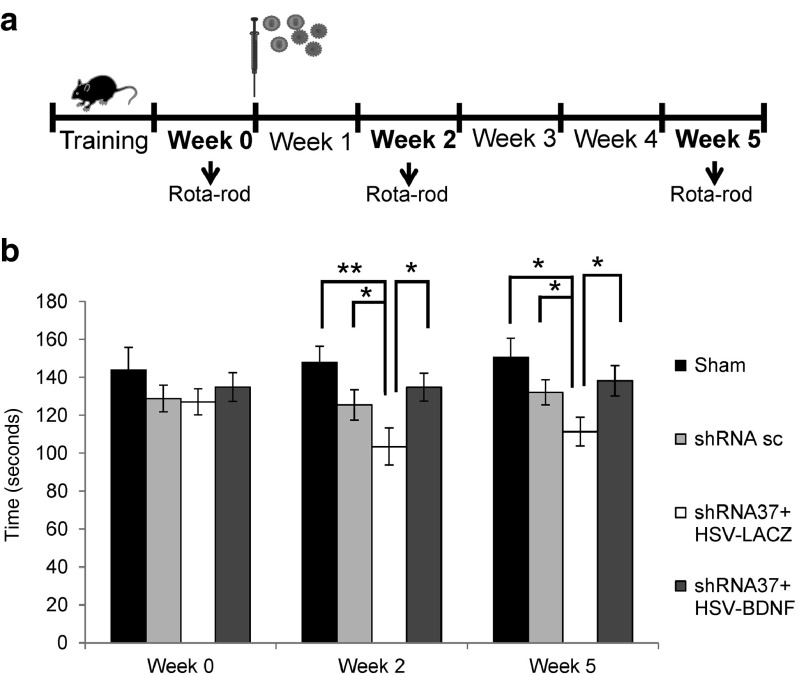
Friedreich's ataxia is a predominantly neurodegenerative disease caused by recessive mutations that produce a deficiency of frataxin (FXN). Here, we have used a herpesviral amplicon vector carrying a gene encoding for brain-derived neurotrophic factor (BDNF) to drive its overexpression in neuronal cells and test for its effect on FXN-deficient neurons both in culture and in the mouse cerebellum in vivo. Gene transfer of BDNF to primary cultures of mouse neurons prevents the apoptosis which is triggered by the knockdown of FXN gene expression. This neuroprotective effect of BDNF is also observed in vivo in a viral vector-based knockdown mouse cerebellar model. The injection of a lentiviral vector carrying a minigene encoding for a FXN-specific short hairpin ribonucleic acid (shRNA) into the mouse cerebellar cortex triggers a FXN deficit which is accompanied by significant apoptosis of granule neurons as well as loss of calbindin in Purkinje cells. These pathological changes are accompanied by a loss of motor coordination of mice as assayed by the rota-rod test. Coinjection of a herpesviral vector encoding for BDNF efficiently prevents both the development of cerebellar neuropathology and the ataxic phenotype. These data demonstrate the potential therapeutic usefulness of neurotrophins like BDNF to protect FXN-deficient neurons from degeneration.
Figures



Comment in
-
Movement disorders: Targeted RNA or BDNF gene transfer protects against frataxin deficiency.Nat Rev Neurol. 2016 Mar;12(3):125. doi: 10.1038/nrneurol.2016.19. Epub 2016 Feb 26. Nat Rev Neurol. 2016. PMID: 26915729 No abstract available.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
