

Therapeutic advances for the tumors associated with neurofibromatosis type 1, type 2, and schwannomatosis
- PMID: 26851632
- PMCID: PMC4827037
- DOI: 10.1093/neuonc/nov200
Therapeutic advances for the tumors associated with neurofibromatosis type 1, type 2, and schwannomatosis
Abstract
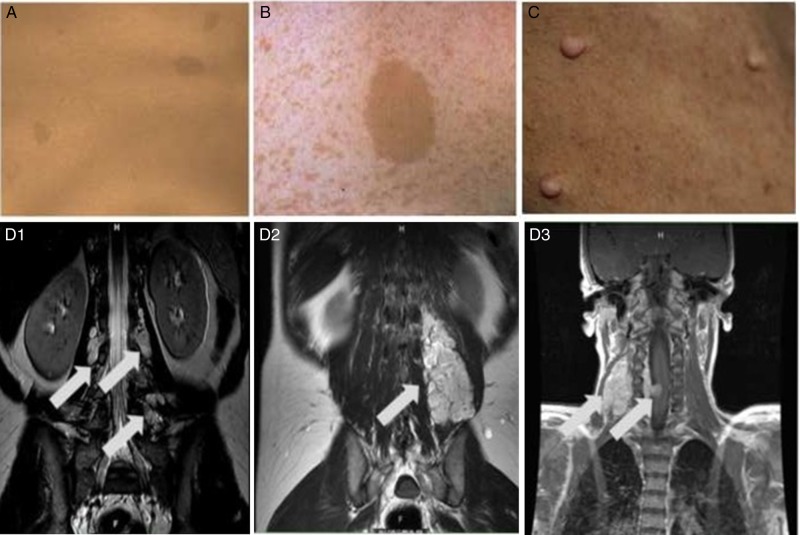
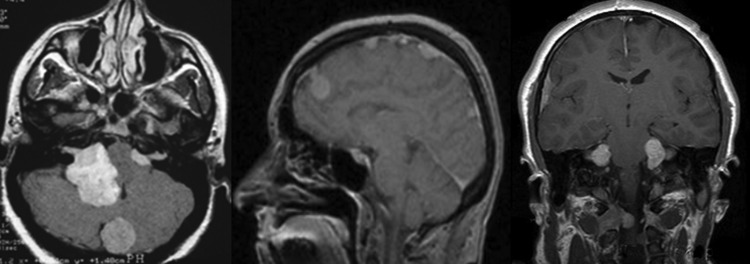
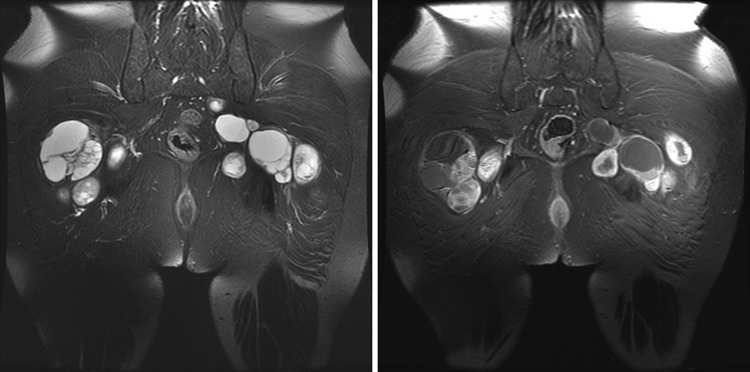
Neurofibromatosis type 1 (NF1), neurofibromatosis type 2 (NF2), and schwannomatosis (SWN) are tumor-suppressor syndromes. Each syndrome is an orphan disease; however, the tumors that arise within them represent the most common tumors of the nervous system worldwide. Systematic investigation of the pathways impacted by the loss of function of neurofibromin (encoded byNF1) and merlin (encoded byNF2) have led to therapeutic advances for patients with NF1 and NF2. In the syndrome of SWN, the genetic landscape is more complex, with 2 known causative genes (SMARCB1andLZTR1) accounting for up to 50% of familial SWN patients. The understanding of the molecular underpinnings of these syndromes is developing rapidly and offers more therapeutic options for the patients. In addition, common sporadic cancers harbor somatic alterations inNF1(ie, glioblastoma, breast cancer, melanoma),NF2(ie, meningioma, mesothelioma) andSMARCB1(ie, atypical teratoid/rhabdoid tumors) such that advances in management of syndromic tumors may benefit patients both with and without germline mutations. In this review, we discuss the clinical and genetic features of NF1, NF2 and SWN, the therapeutic advances for the tumors that arise within these syndromes and the interaction between these rare tumor syndromes and the common tumors that share these mutations.
Keywords: NF1; NF2; schwannomatosis; therapeutics; tumor suppressor syndrome.
© The Author(s) 2016. Published by Oxford University Press on behalf of the Society for Neuro-Oncology. All rights reserved. For permissions, please e-mail: journals.permissions@oup.com.
Figures
References
-
- McBride KA, Ballinger ML, Killick E et al. Li-Fraumeni syndrome: cancer risk assessment and clinical management. Nat Rev Clin Oncol. 2014;11(5):260–271. - PubMed
-
- Huson SM, Harper PS, Compston DA. Von Recklinghausen neurofibromatosis. A clinical and population study in south-east Wales. Brain. 1988;111(Pt 6):1355–1381. - PubMed
-
- Evans DG, Howard E, Giblin C et al. Birth incidence and prevalence of tumor-prone syndromes: estimates from a UK family genetic register service. Am J Med Genet A. 2010;152A(2):327–332. - PubMed
Publication types
MeSH terms
Supplementary concepts
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
