

Method to represent the distribution of QTL additive and dominance effects associated with quantitative traits in computer simulation
- PMID: 26852240
- PMCID: PMC4744427
- DOI: 10.1186/s12859-016-0906-z
Method to represent the distribution of QTL additive and dominance effects associated with quantitative traits in computer simulation
Abstract
Background: Computer simulation is a resource which can be employed to identify optimal breeding strategies to effectively and efficiently achieve specific goals in developing improved cultivars. In some instances, it is crucial to assess in silico the options as well as the impact of various crossing schemes and breeding approaches on performance for traits of interest such as grain yield. For this, a means by which gene effects can be represented in the genome model is critical.
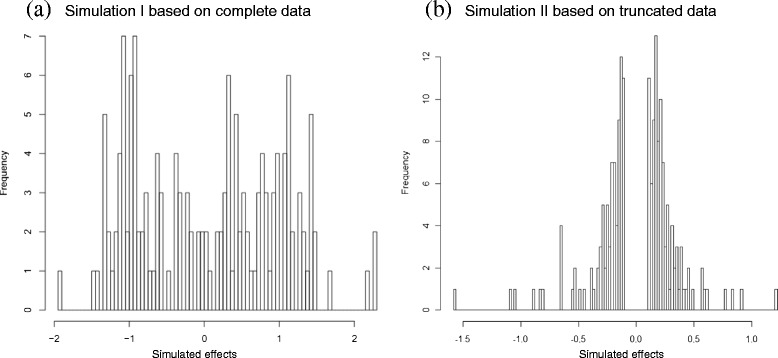
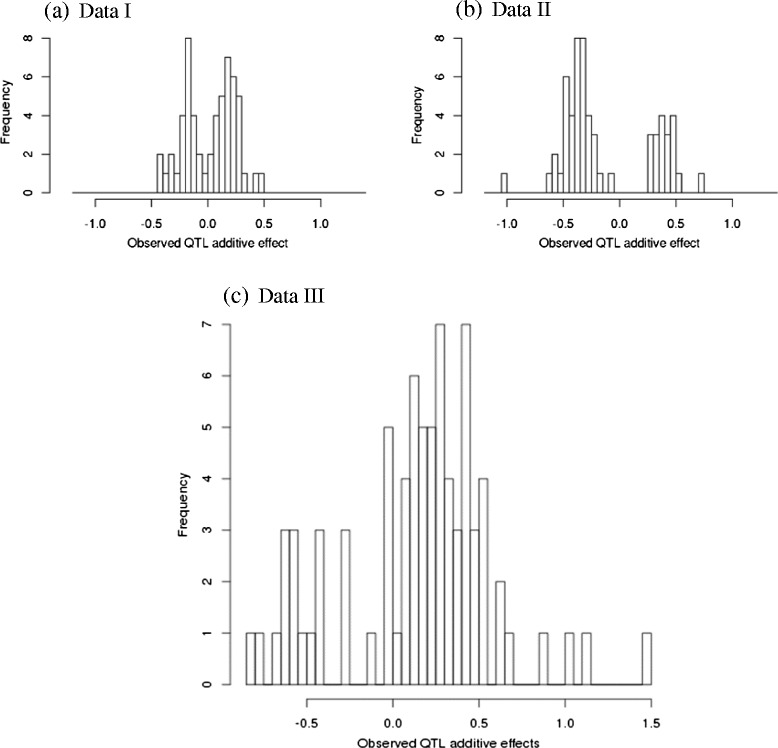
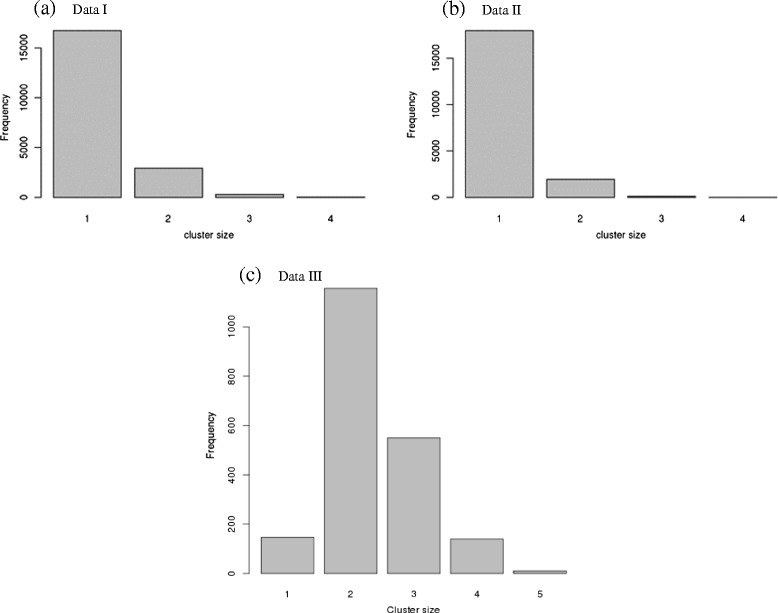
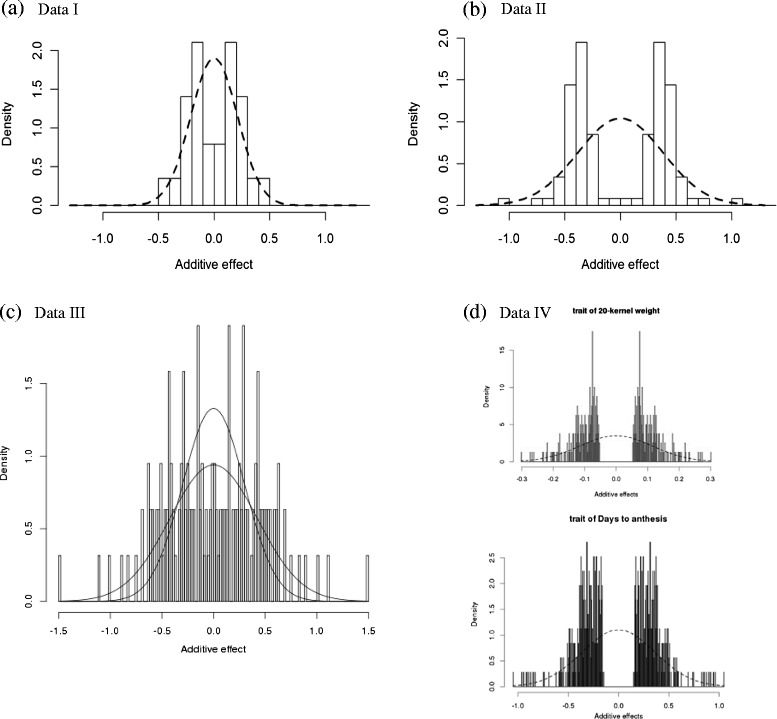
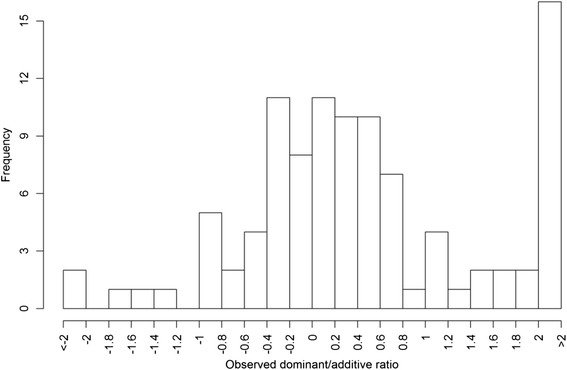
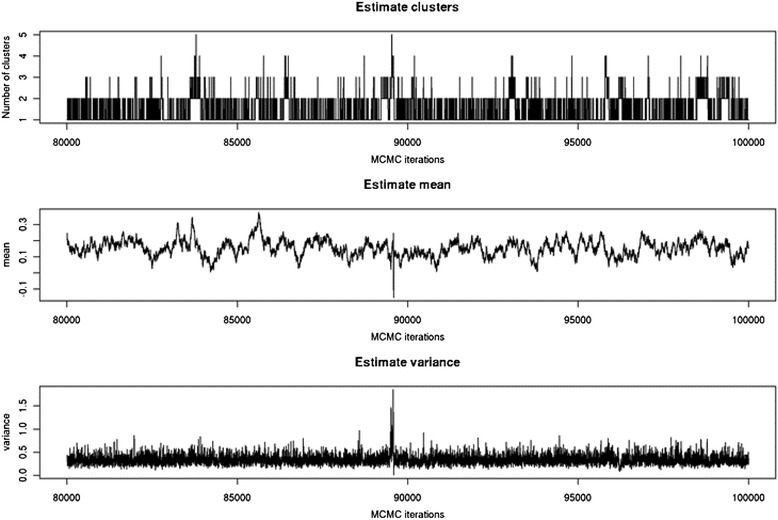
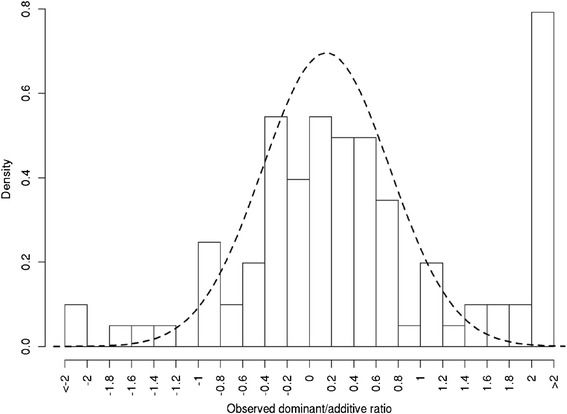
Results: To address this need, we devised a method to represent the genomic distribution of additive and dominance gene effects associated with quantitative traits. The method, based on meta-analysis of previously-estimated QTL effects following Bennewitz and Meuwissen (J Anim Breed Genet 127:171-9, 2010), utilizes a modified Dirichlet process Gaussian mixture model (DPGMM) to fit the number of mixture components and estimate parameters (i.e. mean and variance) of the genomic distribution. The method was demonstrated using several maize QTL data sets to provide estimates of additive and dominance effects for grain yield and other quantitative traits for application in maize genome simulations.
Conclusions: The DPGMM method offers an alternative to the over-simplified infinitesimal model in computer simulation as a means to better represent the genetic architecture of quantitative traits, which likely involve some large effects in addition to many small effects. Furthermore, it confers an advantage over other methods in that the number of mixture model components need not be known a priori. In addition, the method is robust with use of large-scale, multi-allelic data sets or with meta-analyses of smaller QTL data sets which may be derived from bi-parental populations in precisely estimating distribution parameters. Thus, the method has high utility in representing the genetic architecture of quantitative traits in computer simulation.
Figures
Similar articles
-
Genome-wide meta-analysis of maize heterosis reveals the potential role of additive gene expression at pericentromeric loci.BMC Plant Biol. 2014 Apr 2;14:88. doi: 10.1186/1471-2229-14-88. BMC Plant Biol. 2014. PMID: 24693880 Free PMC article.
-
Yield QTLome distribution correlates with gene density in maize.Plant Sci. 2016 Jan;242:300-309. doi: 10.1016/j.plantsci.2015.09.022. Epub 2015 Sep 28. Plant Sci. 2016. PMID: 26566847
-
QTL Mapping of Kernel Number-Related Traits and Validation of One Major QTL for Ear Length in Maize.PLoS One. 2016 May 13;11(5):e0155506. doi: 10.1371/journal.pone.0155506. eCollection 2016. PLoS One. 2016. PMID: 27176215 Free PMC article.
-
Mapping QTL for agronomic traits in breeding populations.Theor Appl Genet. 2012 Jul;125(2):201-10. doi: 10.1007/s00122-012-1887-6. Epub 2012 May 22. Theor Appl Genet. 2012. PMID: 22614179 Review.
-
Methodologies for segregation analysis and QTL mapping in plants.Genetica. 2009 Jun;136(2):311-8. doi: 10.1007/s10709-008-9313-3. Epub 2008 Aug 23. Genetica. 2009. PMID: 18726162 Review.
Cited by
-
A look-ahead Monte Carlo simulation method for improving parental selection in trait introgression.Sci Rep. 2021 Feb 16;11(1):3918. doi: 10.1038/s41598-021-83634-x. Sci Rep. 2021. PMID: 33594238 Free PMC article.
-
Predicting the purebred-crossbred genetic correlation from the genetic variance components in the parental lines.Genet Sel Evol. 2021 Feb 4;53(1):10. doi: 10.1186/s12711-021-00601-w. Genet Sel Evol. 2021. PMID: 33541267 Free PMC article.
-
Optimized breeding strategies for multiple trait integration: III. Parameters for success in version testing.Mol Breed. 2015;35(10):201. doi: 10.1007/s11032-015-0397-z. Epub 2015 Oct 14. Mol Breed. 2015. PMID: 26491398 Free PMC article.
-
The long-term effects of genomic selection: 1. Response to selection, additive genetic variance, and genetic architecture.Genet Sel Evol. 2022 Mar 7;54(1):19. doi: 10.1186/s12711-022-00709-7. Genet Sel Evol. 2022. PMID: 35255802 Free PMC article.
-
The Impact of Non-additive Effects on the Genetic Correlation Between Populations.G3 (Bethesda). 2020 Feb 6;10(2):783-795. doi: 10.1534/g3.119.400663. G3 (Bethesda). 2020. PMID: 31857332 Free PMC article.
References
-
- Sun X, Peng T, Mumm RH. The role and basics of computer simulation in support of critical decisions in plant breeding. Mol Breed. 2011;28(4):421–436. doi: 10.1007/s11032-011-9630-6. - DOI
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
