

Secreted IGFBP5 mediates mTORC1-dependent feedback inhibition of IGF-1 signalling
- PMID: 26854565
- PMCID: PMC5001501
- DOI: 10.1038/ncb3311
Secreted IGFBP5 mediates mTORC1-dependent feedback inhibition of IGF-1 signalling
Abstract
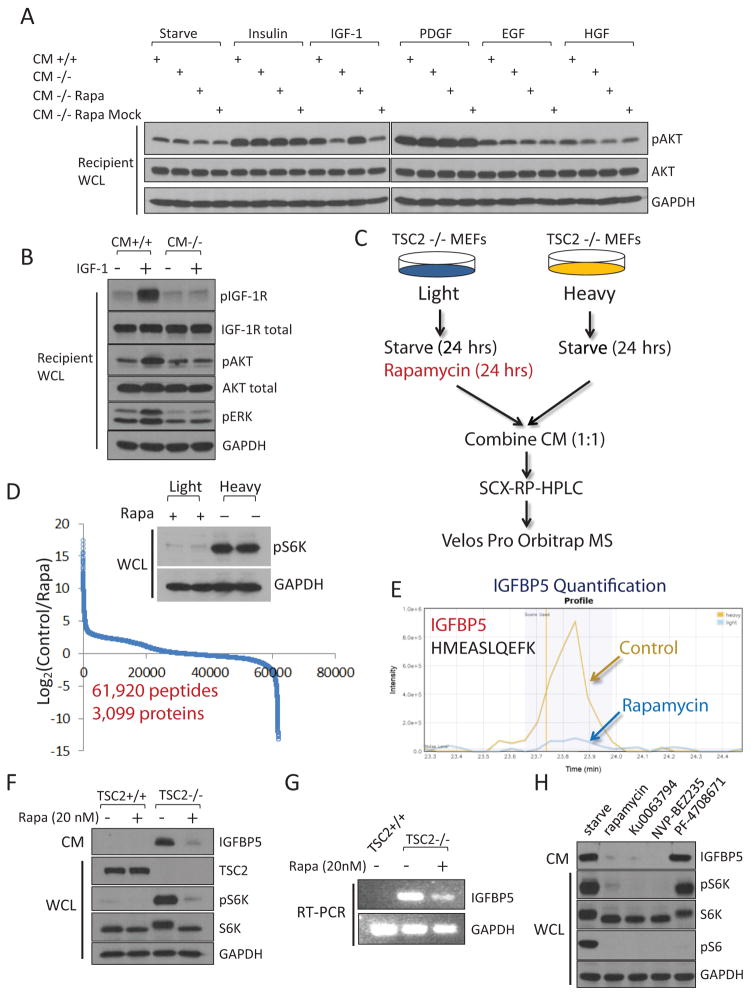
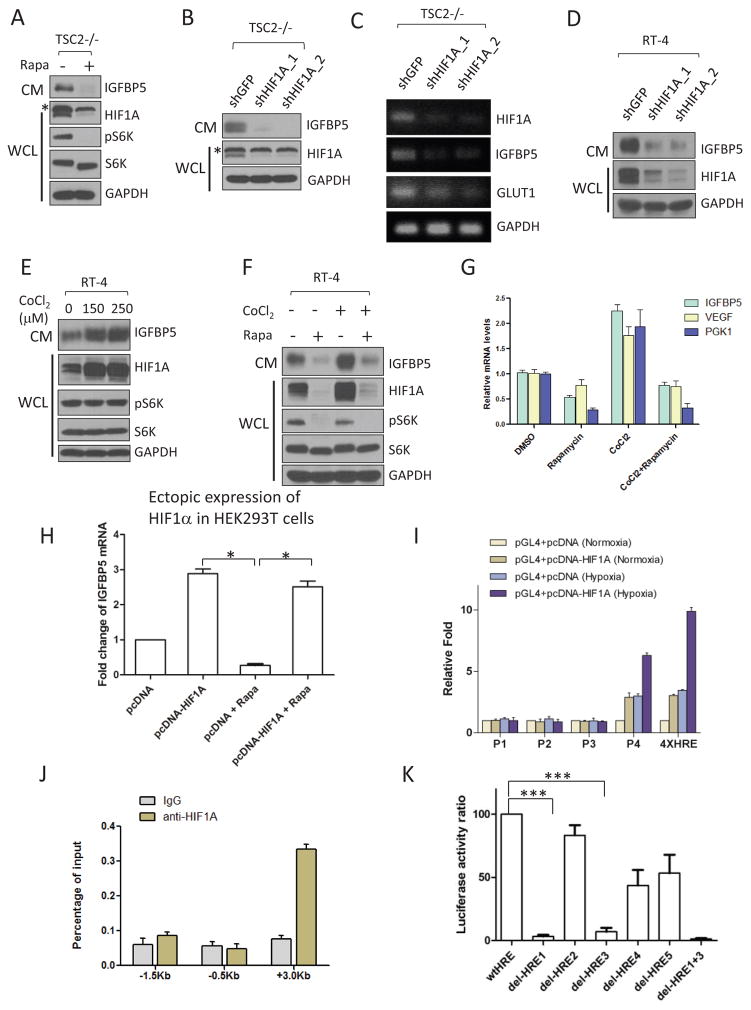
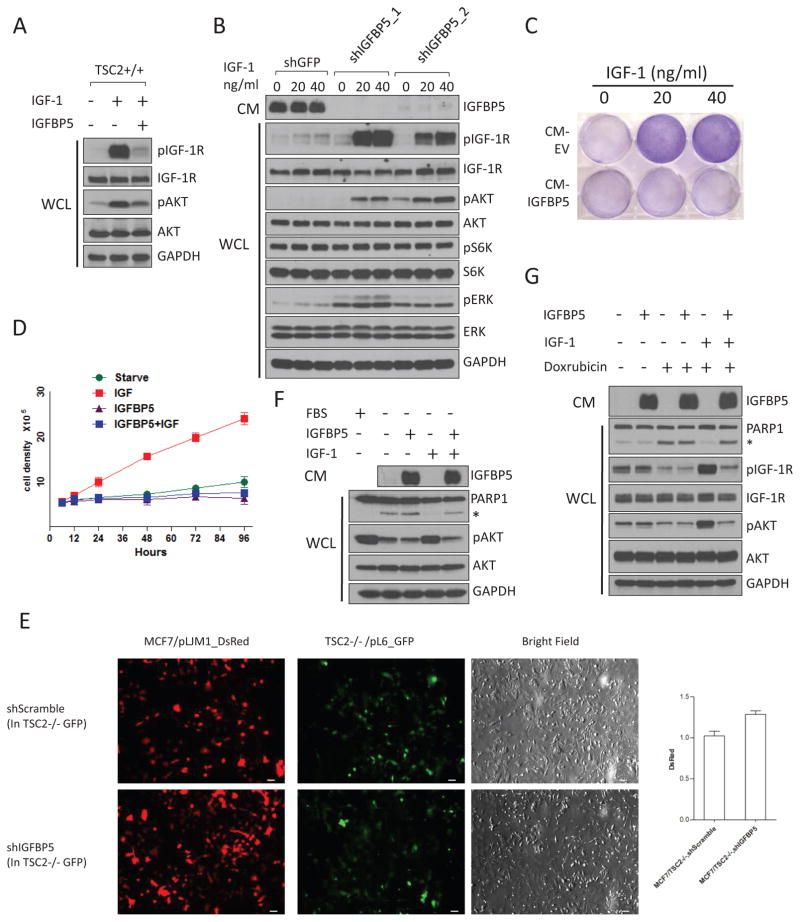
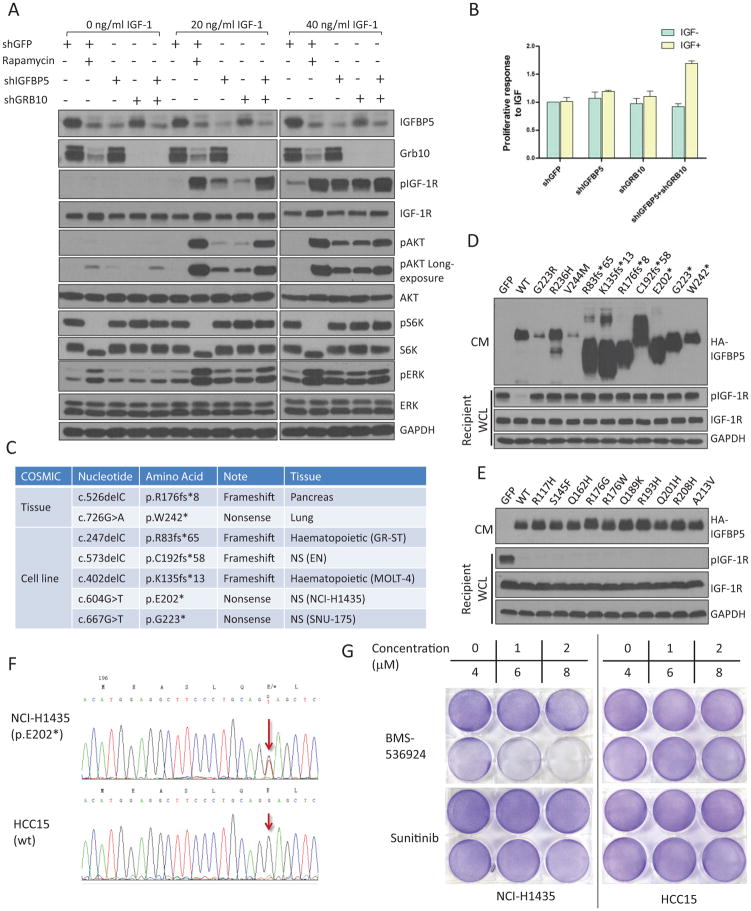
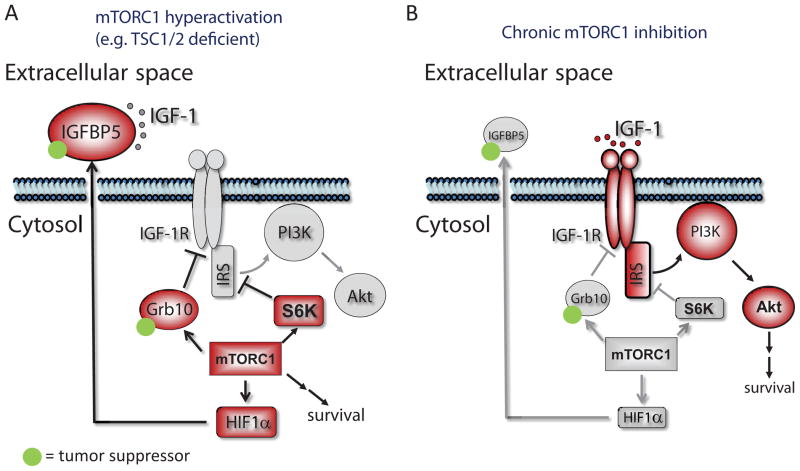
The PI(3)K-Akt-mTORC1 pathway is a highly dynamic network that is balanced and stabilized by a number of feedback inhibition loops. Specifically, activation of mTORC1 has been shown to lead to the inhibition of its upstream growth factor signalling. Activation of the growth factor receptors is triggered by the binding of their cognate ligands in the extracellular space. However, whether secreted proteins contribute to the mTORC1-dependent feedback loops remains unclear. We found that cells with hyperactive mTORC1 secrete a protein that potently inhibits the function of IGF-1. Using a large-scale, unbiased quantitative proteomic platform, we comprehensively characterized the rapamycin-sensitive secretome in TSC2(-/-) mouse embryonic fibroblasts, and identified IGFBP5 as a secreted, mTORC1 downstream effector protein. IGFBP5 is a direct transcriptional target of HIF1, which itself is a known mTORC1 target. IGFBP5 is a potent inhibitor of both the signalling and functional outputs of IGF-1. Once secreted, IGFBP5 cooperates with intracellular branches of the feedback mechanisms to block the activation of IGF-1 signalling. Finally, IGFBP5 is a potential tumour suppressor, and the proliferation of IGFBP5-mutated cancer cells is selectively blocked by IGF-1R inhibitors.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
