

Integrative approaches for large-scale transcriptome-wide association studies
- PMID: 26854917
- PMCID: PMC4767558
- DOI: 10.1038/ng.3506
Integrative approaches for large-scale transcriptome-wide association studies
Abstract
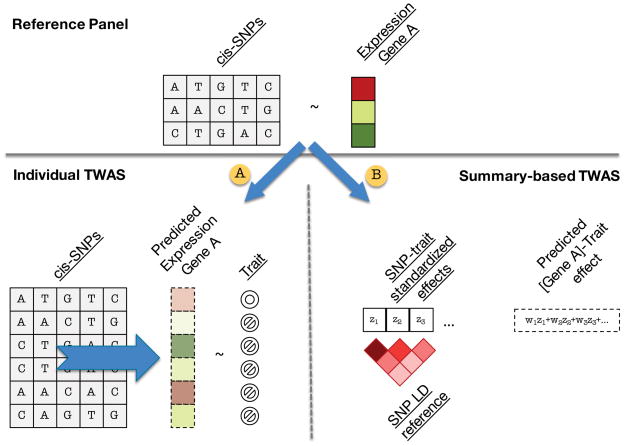
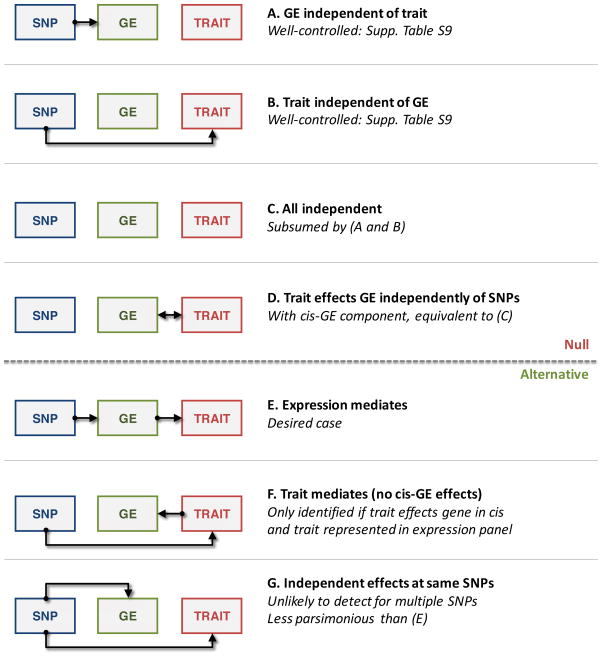
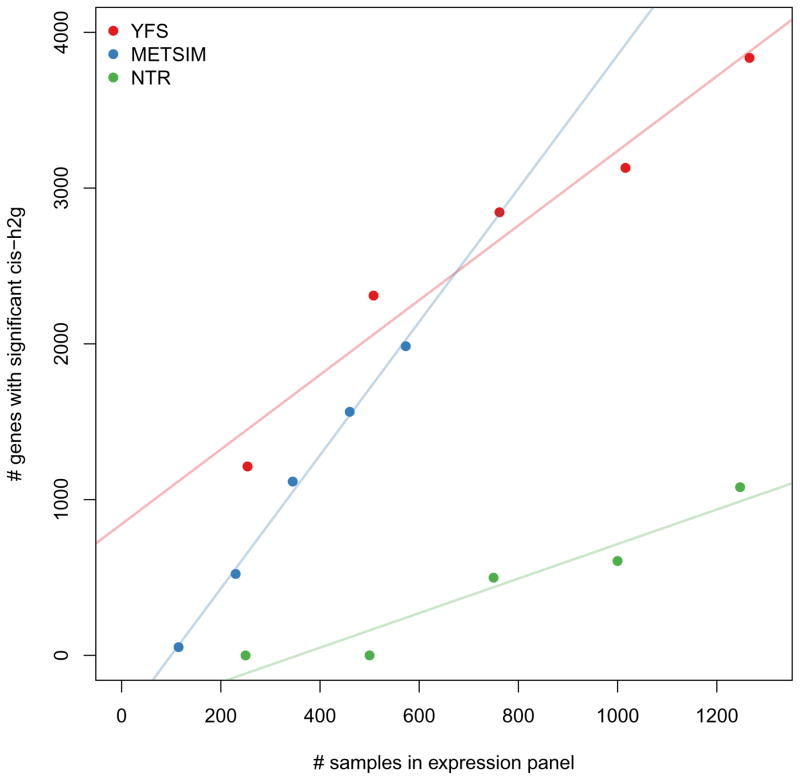
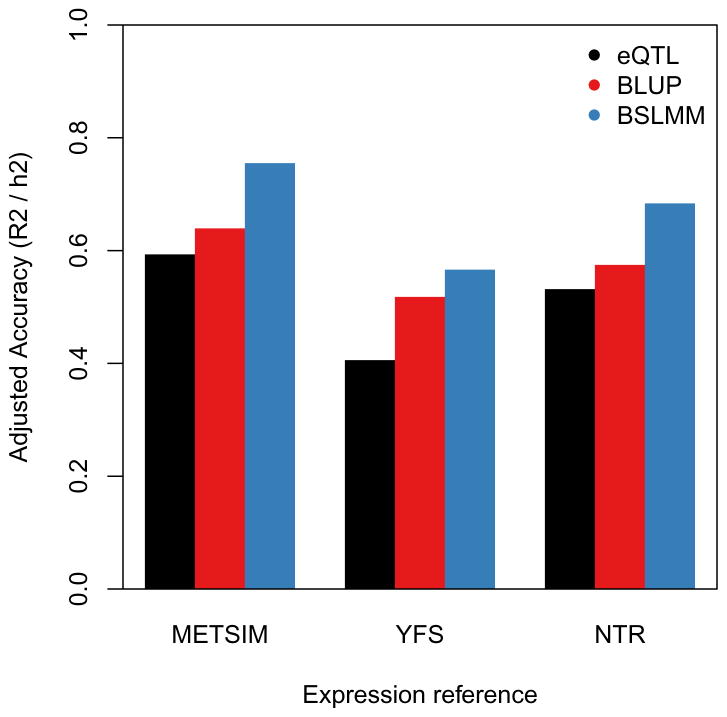
Many genetic variants influence complex traits by modulating gene expression, thus altering the abundance of one or multiple proteins. Here we introduce a powerful strategy that integrates gene expression measurements with summary association statistics from large-scale genome-wide association studies (GWAS) to identify genes whose cis-regulated expression is associated with complex traits. We leverage expression imputation from genetic data to perform a transcriptome-wide association study (TWAS) to identify significant expression-trait associations. We applied our approaches to expression data from blood and adipose tissue measured in ∼ 3,000 individuals overall. We imputed gene expression into GWAS data from over 900,000 phenotype measurements to identify 69 new genes significantly associated with obesity-related traits (BMI, lipids and height). Many of these genes are associated with relevant phenotypes in the Hybrid Mouse Diversity Panel. Our results showcase the power of integrating genotype, gene expression and phenotype to gain insights into the genetic basis of complex traits.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
Comment in
-
Complex traits: Integrating gene variation and expression to understand complex traits.Nat Rev Genet. 2016 Apr;17(4):194. doi: 10.1038/nrg.2016.18. Epub 2016 Feb 22. Nat Rev Genet. 2016. PMID: 26900024 No abstract available.
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
