

CYP3A5 mediates basal and acquired therapy resistance in different subtypes of pancreatic ductal adenocarcinoma
- PMID: 26855150
- PMCID: PMC4780258
- DOI: 10.1038/nm.4038
CYP3A5 mediates basal and acquired therapy resistance in different subtypes of pancreatic ductal adenocarcinoma
Abstract
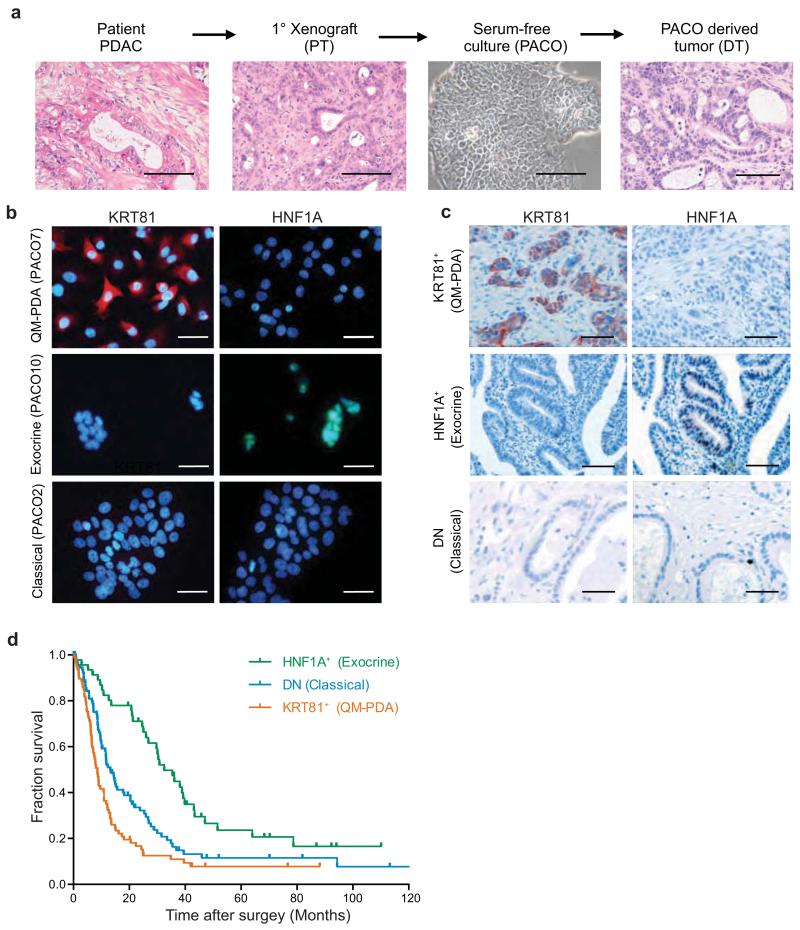
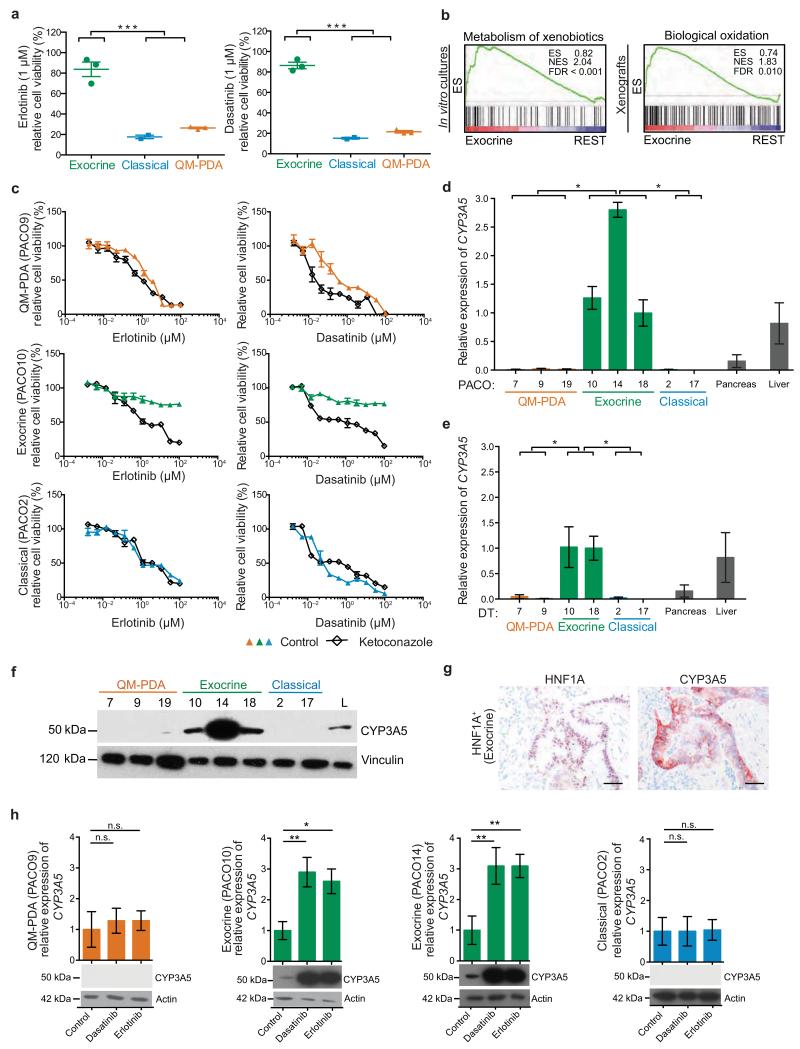
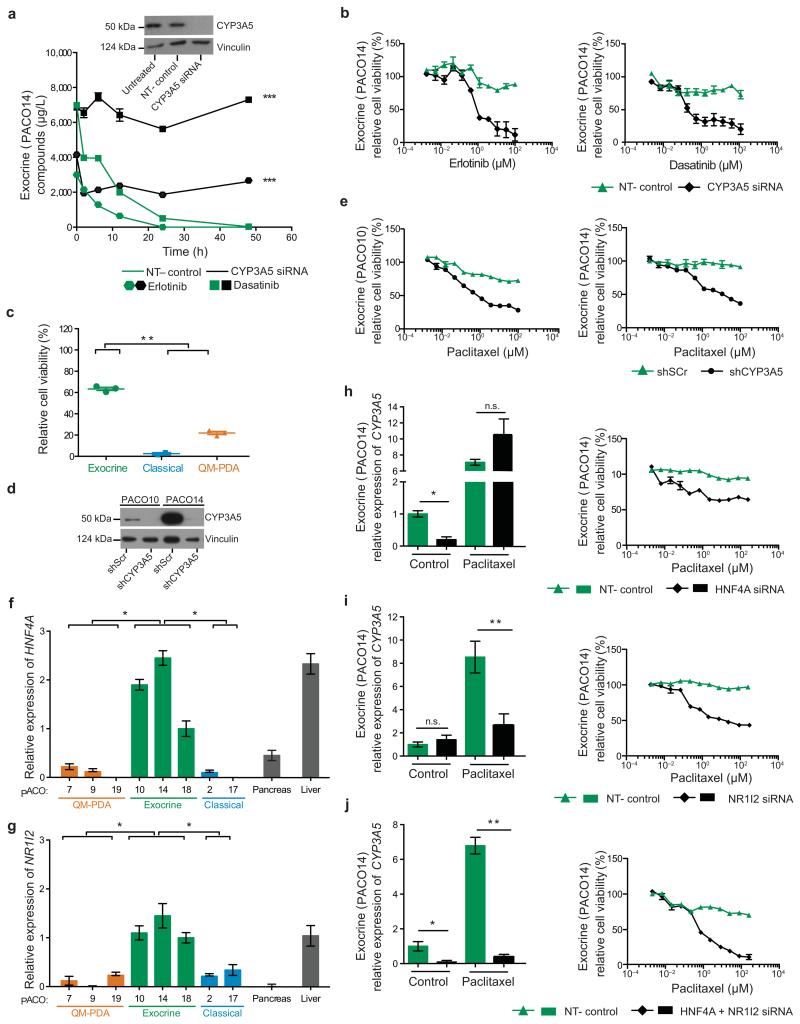
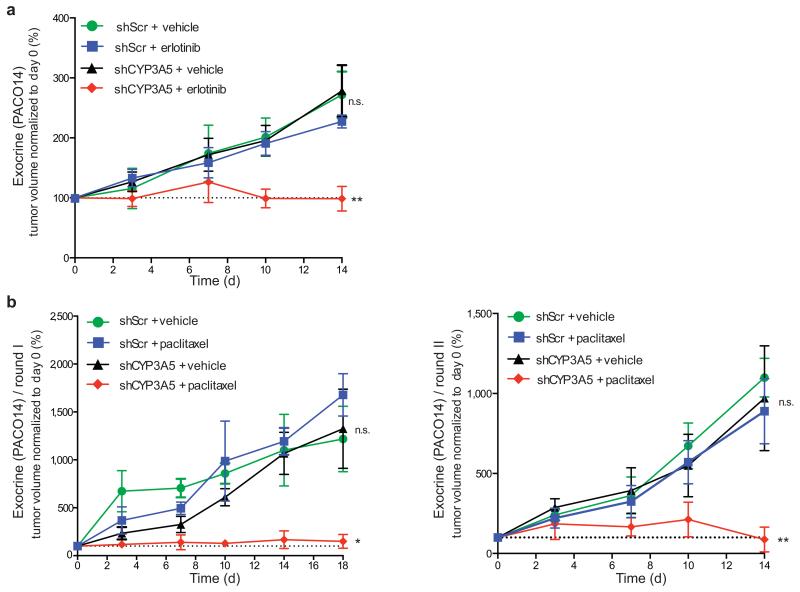
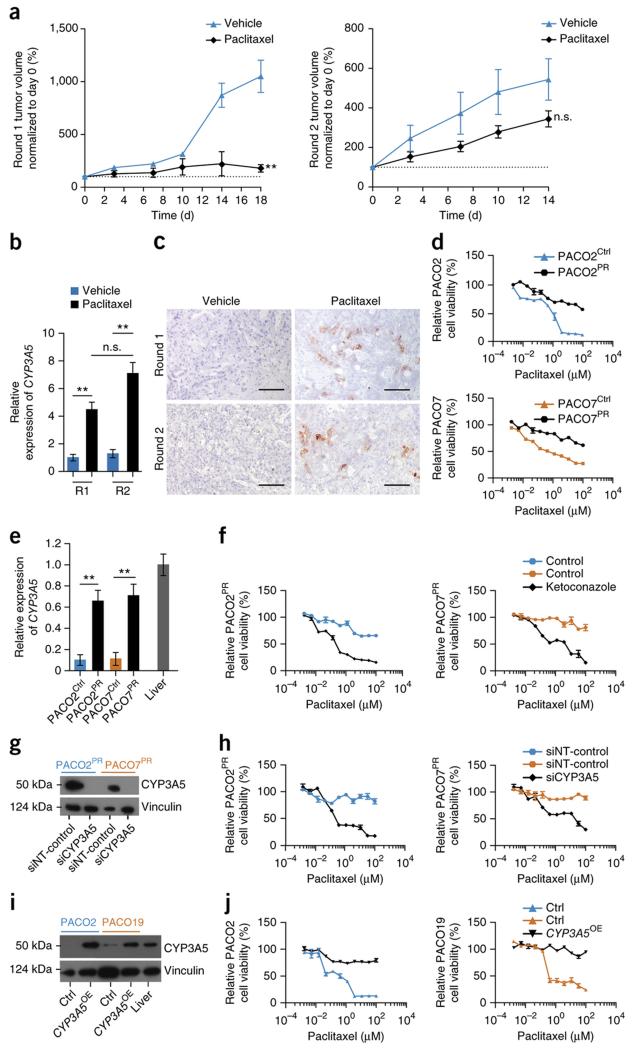
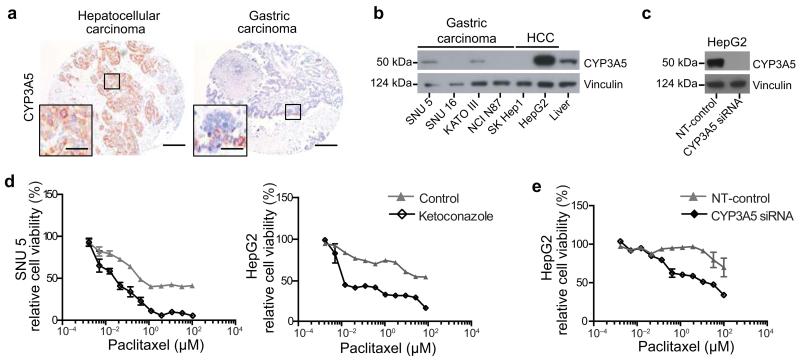
Although subtypes of pancreatic ductal adenocarcinoma (PDAC) have been described, this malignancy is clinically still treated as a single disease. Here we present patient-derived models representing the full spectrum of previously identified quasi-mesenchymal (QM-PDA), classical and exocrine-like PDAC subtypes, and identify two markers--HNF1A and KRT81--that enable stratification of tumors into different subtypes by using immunohistochemistry. Individuals with tumors of these subtypes showed substantial differences in overall survival, and their tumors differed in drug sensitivity, with the exocrine-like subtype being resistant to tyrosine kinase inhibitors and paclitaxel. Cytochrome P450 3A5 (CYP3A5) metabolizes these compounds in tumors of the exocrine-like subtype, and pharmacological or short hairpin RNA (shRNA)-mediated CYP3A5 inhibition sensitizes tumor cells to these drugs. Whereas hepatocyte nuclear factor 4, alpha (HNF4A) controls basal expression of CYP3A5, drug-induced CYP3A5 upregulation is mediated by the nuclear receptor NR1I2. CYP3A5 also contributes to acquired drug resistance in QM-PDA and classical PDAC, and it is highly expressed in several additional malignancies. These findings designate CYP3A5 as a predictor of therapy response and as a tumor cell-autonomous detoxification mechanism that must be overcome to prevent drug resistance.
Figures
Comment in
-
Pancreatic cancer: CYP3A5 contributes to PDAC chemoresistance.Nat Rev Gastroenterol Hepatol. 2016 Apr;13(4):188. doi: 10.1038/nrgastro.2016.36. Epub 2016 Feb 24. Nat Rev Gastroenterol Hepatol. 2016. PMID: 26907883 No abstract available.
-
HNF1A, KRT81, and CYP3A5: three more straws on the back of pancreatic cancer?Transl Cancer Res. 2016 Aug;5(Suppl 2):S253-S256. doi: 10.21037/tcr.2016.08.12. Transl Cancer Res. 2016. PMID: 30581769 Free PMC article. No abstract available.
References
-
- Hidalgo M. Pancreatic cancer. The New England journal of medicine. 2010;362:1605–1617. - PubMed
-
- Malvezzi M, Bertuccio P, Levi F, La Vecchia C, Negri E. European cancer mortality predictions for the year 2014. Annals of oncology : official journal of the European Society for Medical Oncology / ESMO. 2014 - PubMed
-
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA: a cancer journal for clinicians. 2015;65:5–29. - PubMed
-
- Burris HA, 3rd, et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: a randomized trial. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 1997;15:2403–2413. - PubMed
-
- Conroy T, et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. The New England journal of medicine. 2011;364:1817–1825. - PubMed
Methods-only references
-
- Huber W, von Heydebreck A, Sultmann H, Poustka A, Vingron M. Variance stabilization applied to microarray data calibration and to the quantification of differential expression. Bioinformatics. 2002;18(Suppl 1):S96–104. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
