

Metformin promotes tau aggregation and exacerbates abnormal behavior in a mouse model of tauopathy
- PMID: 26858121
- PMCID: PMC4746897
- DOI: 10.1186/s13024-016-0082-7
Metformin promotes tau aggregation and exacerbates abnormal behavior in a mouse model of tauopathy
Abstract
Background: Alzheimer disease (AD) and other tauopathies develop cerebral intracellular inclusions of hyperphosphorylated tau. Epidemiological and experimental evidence suggests a clear link between type 2 diabetes mellitus and AD. In AD animal models, tau pathology is exacerbated by metabolic comorbidities, such as insulin resistance and diabetes. Within this context, anitidiabetic drugs, including the widely-prescribed insulin-sensitizing drug metformin, are currently being investigated for AD therapy. However, their efficacy for tauopathy in vivo has not been tested.
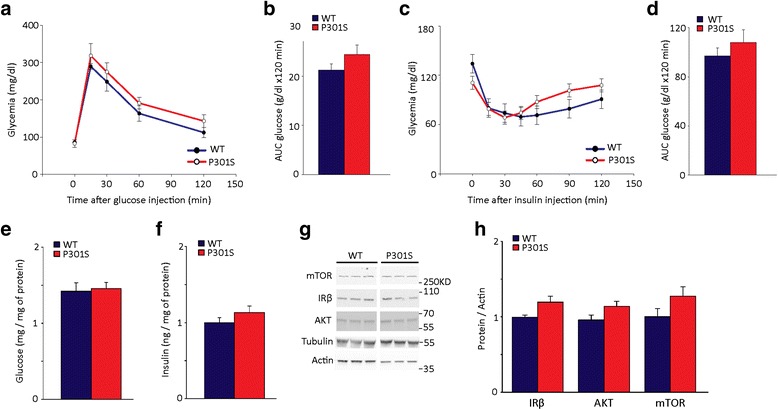
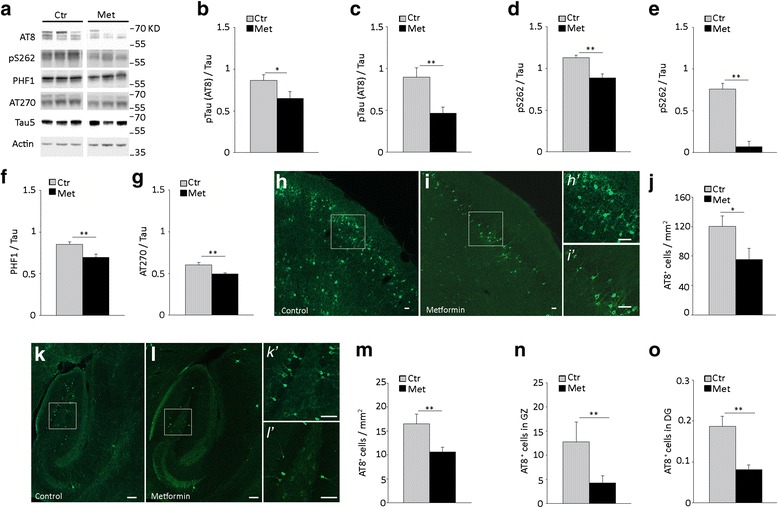
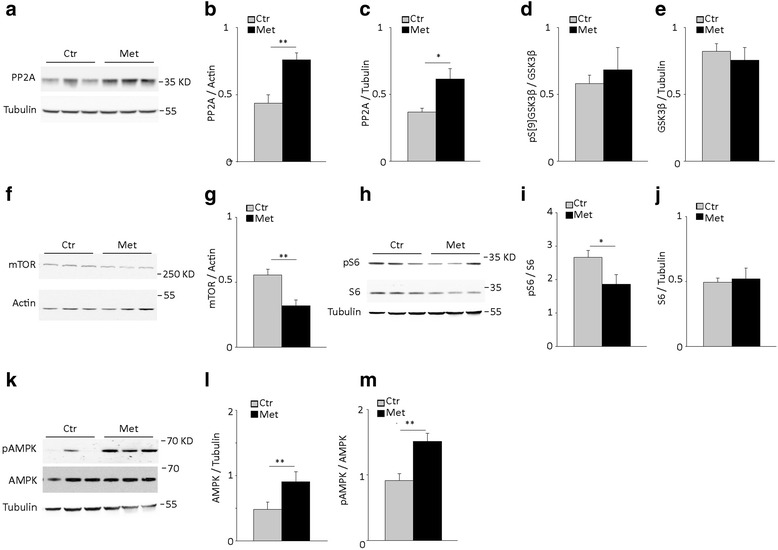
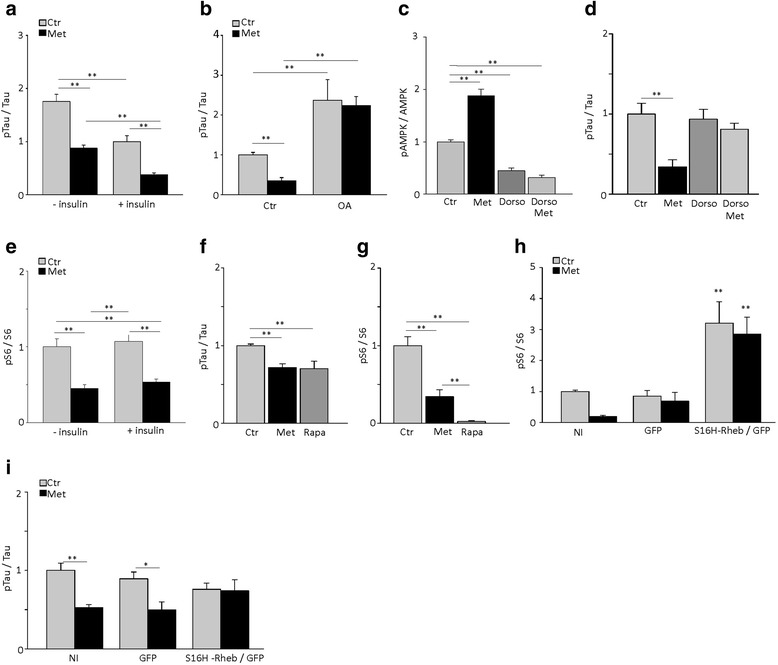
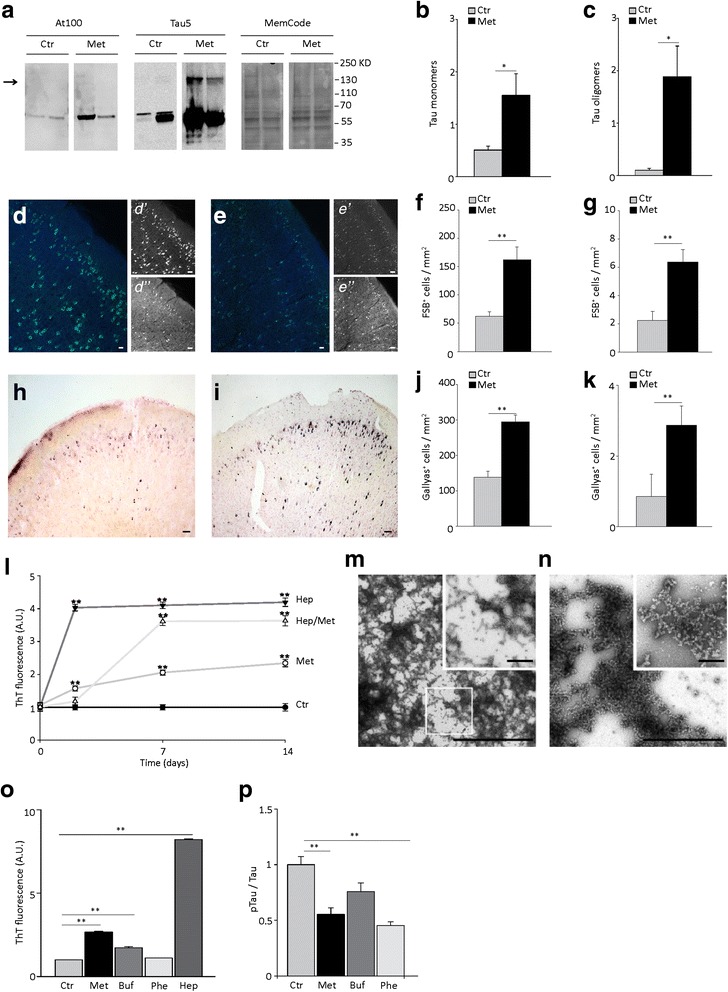
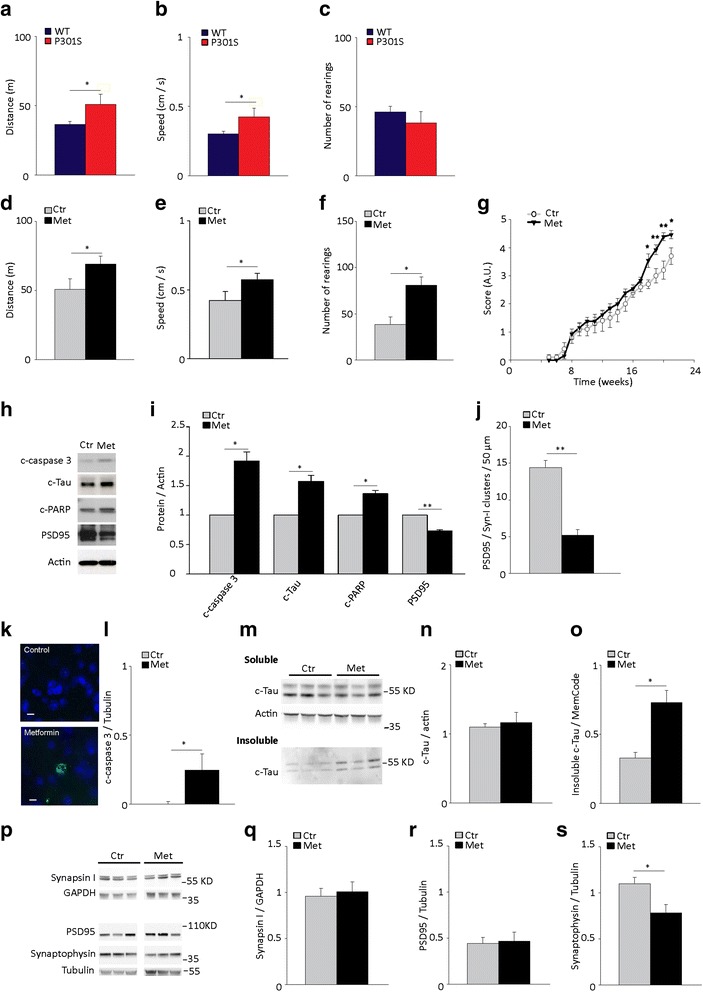
Results: Here, we report that in the P301S mutant human tau (P301S) transgenic mouse model of tauopathy, chronic administration of metformin exerts paradoxical effects on tau pathology. Despite reducing tau phosphorylation in the cortex and hippocampus via AMPK/mTOR and PP2A, metformin increases insoluble tau species (including tau oligomers) and the number of inclusions with β-sheet aggregates in the brain of P301S mice. In addition, metformin exacerbates hindlimb atrophy, increases P301S hyperactive behavior, induces tau cleavage by caspase 3 and disrupts synaptic structures.
Conclusions: These findings indicate that metformin pro-aggregation effects mitigate the potential benefits arising from its dephosphorylating action, possibly leading to an overall increase of the risk of tauopathy in elderly diabetic patients.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
