

Plakophilin-2 loss promotes TGF-β1/p38 MAPK-dependent fibrotic gene expression in cardiomyocytes
- PMID: 26858265
- PMCID: PMC4754716
- DOI: 10.1083/jcb.201507018
Plakophilin-2 loss promotes TGF-β1/p38 MAPK-dependent fibrotic gene expression in cardiomyocytes
Abstract
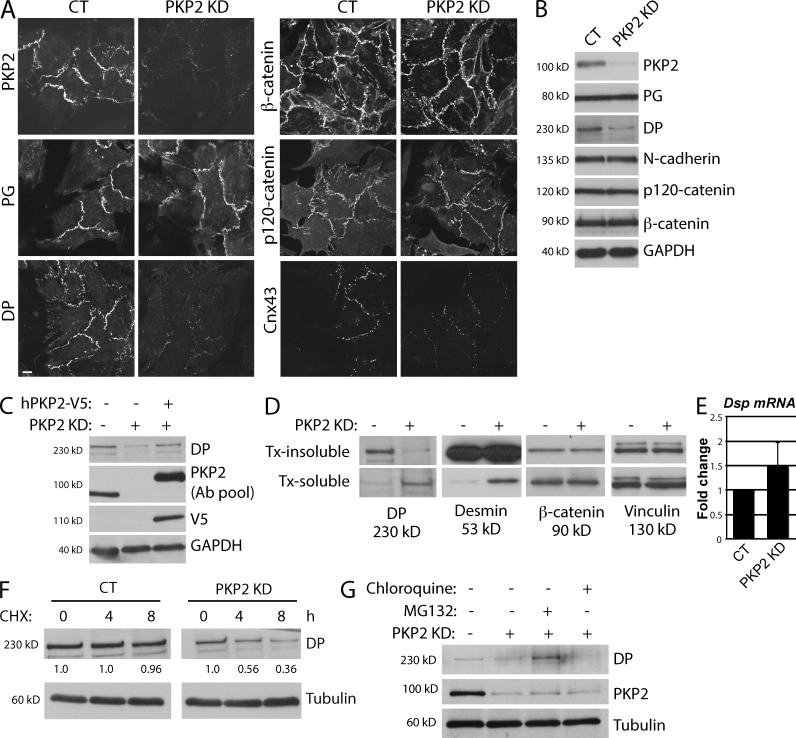
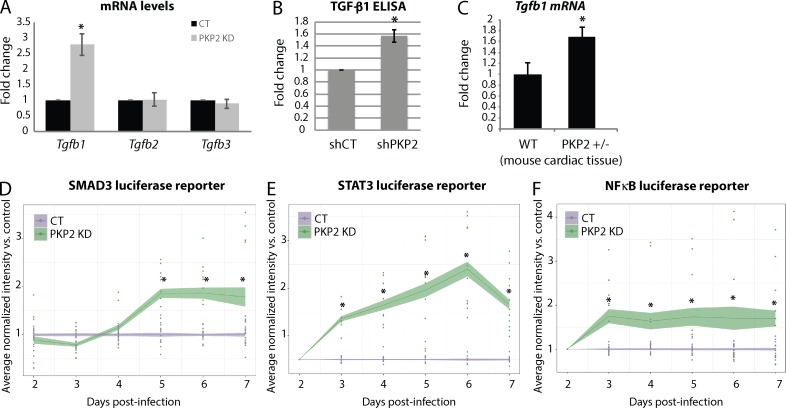
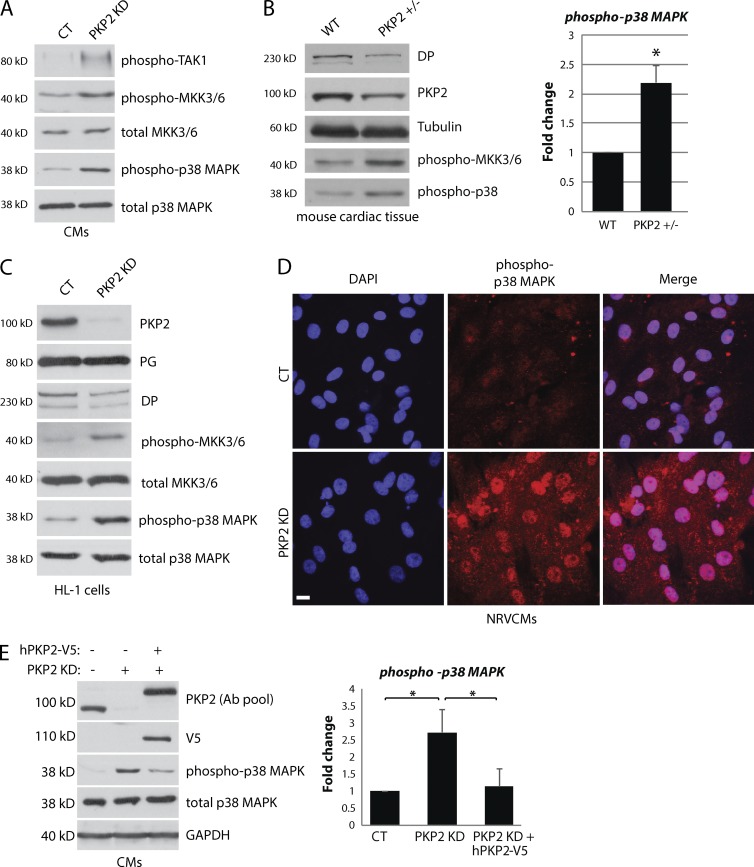
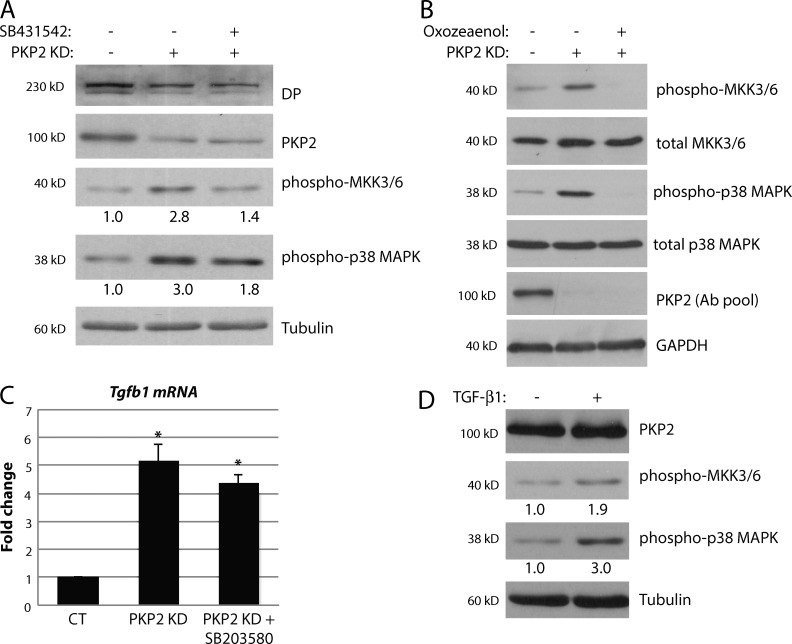
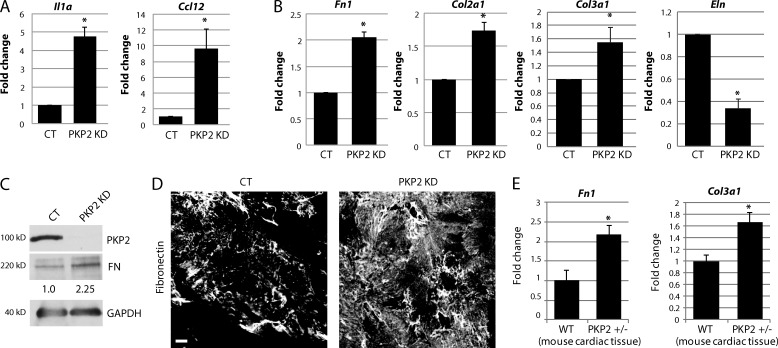
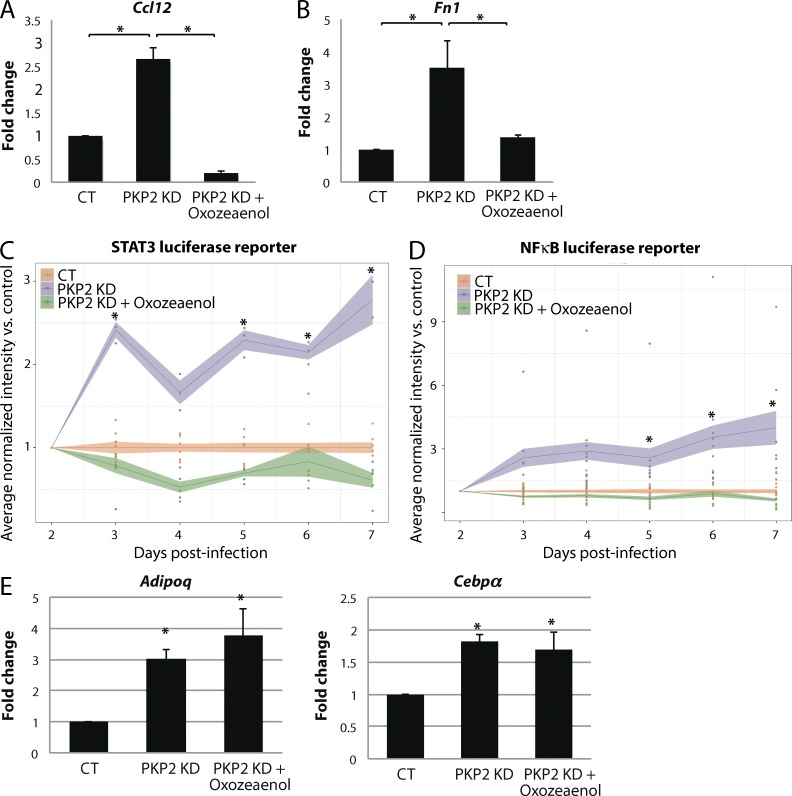
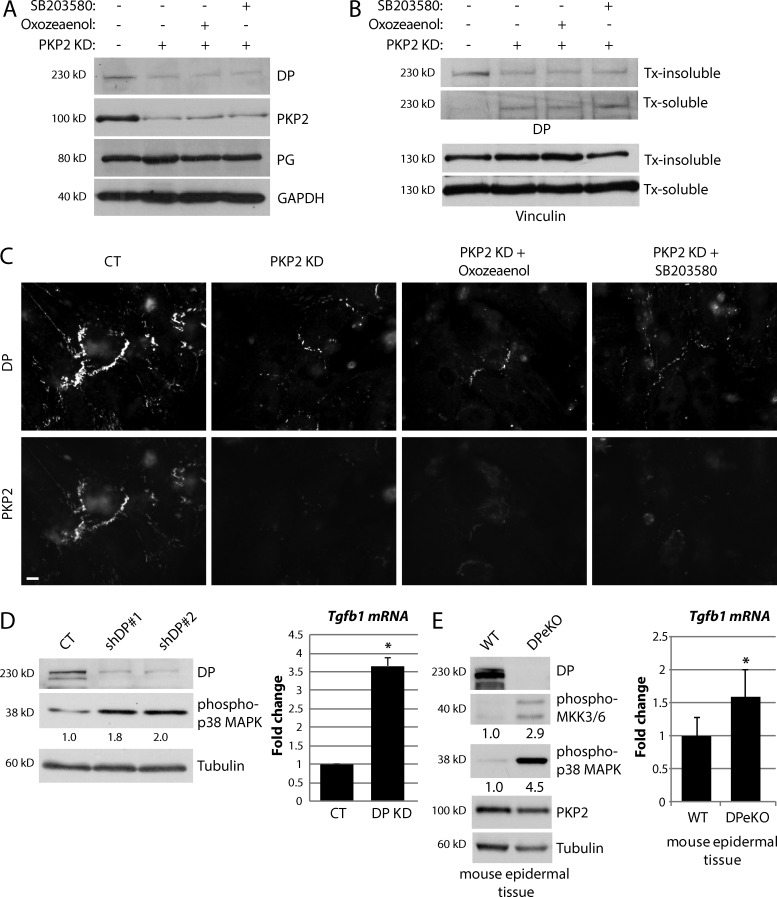
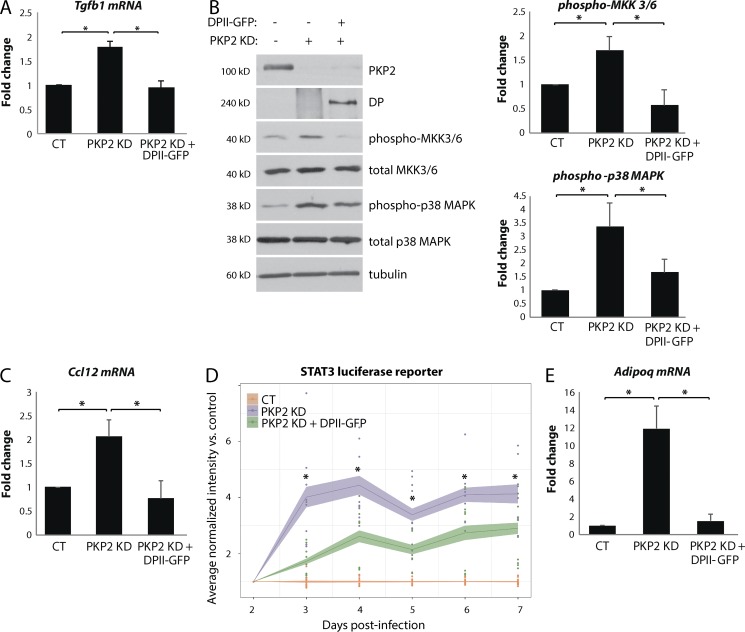
Members of the desmosome protein family are integral components of the cardiac area composita, a mixed junctional complex responsible for electromechanical coupling between cardiomyocytes. In this study, we provide evidence that loss of the desmosomal armadillo protein Plakophilin-2 (PKP2) in cardiomyocytes elevates transforming growth factor β1 (TGF-β1) and p38 mitogen-activated protein kinase (MAPK) signaling, which together coordinate a transcriptional program that results in increased expression of profibrotic genes. Importantly, we demonstrate that expression of Desmoplakin (DP) is lost upon PKP2 knockdown and that restoration of DP expression rescues the activation of this TGF-β1/p38 MAPK transcriptional cascade. Tissues from PKP2 heterozygous and DP conditional knockout mouse models also exhibit elevated TGF-β1/p38 MAPK signaling and induction of fibrotic gene expression in vivo. These data therefore identify PKP2 and DP as central players in coordination of desmosome-dependent TGF-β1/p38 MAPK signaling in cardiomyocytes, pathways known to play a role in different types of cardiac disease, such as arrhythmogenic or hypertrophic cardiomyopathy.
© 2016 Dubash et al.
Figures
References
-
- Angst B.D., Nilles L.A., and Green K.J.. 1990. Desmoplakin II expression is not restricted to stratified epithelia. J. Cell Sci. 97:247–257. - PubMed
-
- Asimaki A., Tandri H., Duffy E.R., Winterfield J.R., Mackey-Bojack S., Picken M.M., Cooper L.T., Wilber D.J., Marcus F.I., Basso C., et al. 2011. Altered desmosomal proteins in granulomatous myocarditis and potential pathogenic links to arrhythmogenic right ventricular cardiomyopathy. Circ Arrhythm Electrophysiol. 4:743–752. 10.1161/CIRCEP.111.964890 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- R37 AR043380/AR/NIAMS NIH HHS/United States
- R01 AR043380/AR/NIAMS NIH HHS/United States
- GM057691/GM/NIGMS NIH HHS/United States
- R01 GM057691/GM/NIGMS NIH HHS/United States
- P30 CA060553/CA/NCI NIH HHS/United States
- P30CA060553/CA/NCI NIH HHS/United States
- AR43380/AR/NIAMS NIH HHS/United States
- R01 CA122151/CA/NCI NIH HHS/United States
- R01 AR041836/AR/NIAMS NIH HHS/United States
- HL106632/HL/NHLBI NIH HHS/United States
- R01 CA173745/CA/NCI NIH HHS/United States
- T32 GM008152/GM/NIGMS NIH HHS/United States
- R01 HL106632/HL/NHLBI NIH HHS/United States
- CA122151/CA/NCI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
