

Genome-wide prediction models that incorporate de novo GWAS are a powerful new tool for tropical rice improvement
- PMID: 26860200
- PMCID: PMC4806696
- DOI: 10.1038/hdy.2015.113
Genome-wide prediction models that incorporate de novo GWAS are a powerful new tool for tropical rice improvement
Abstract
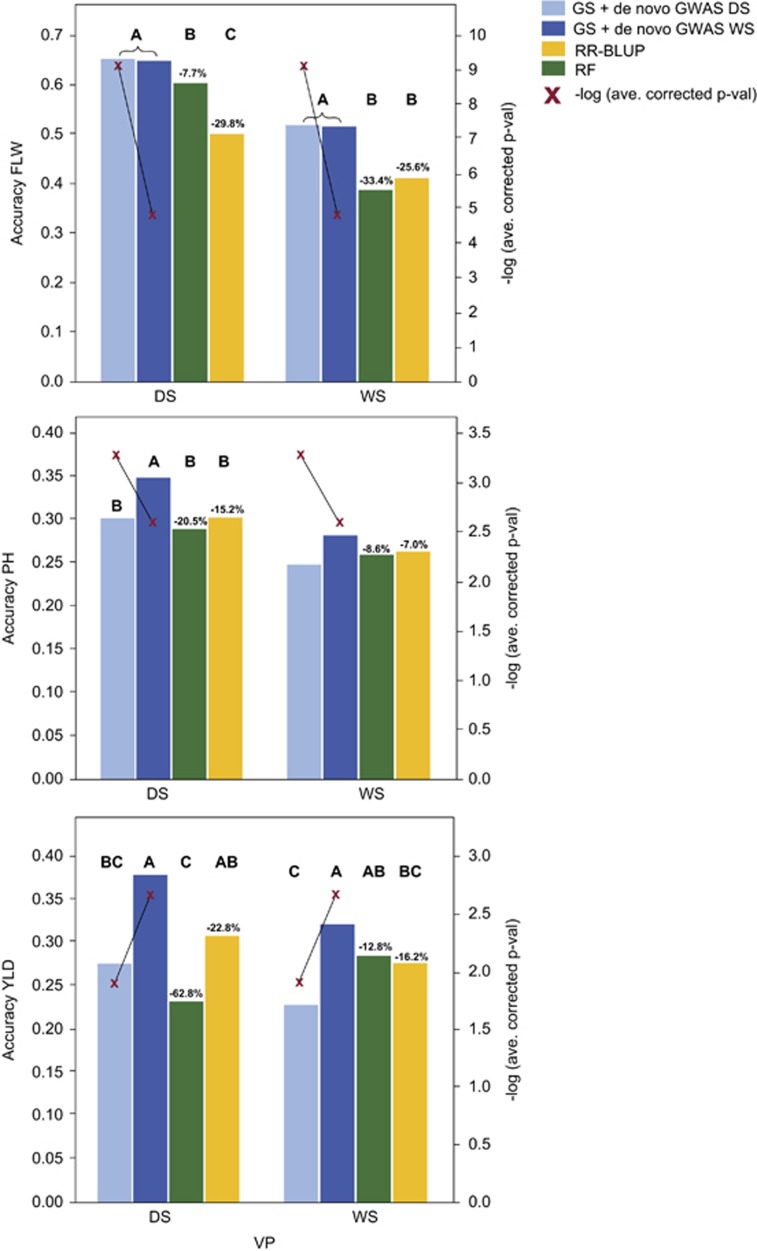
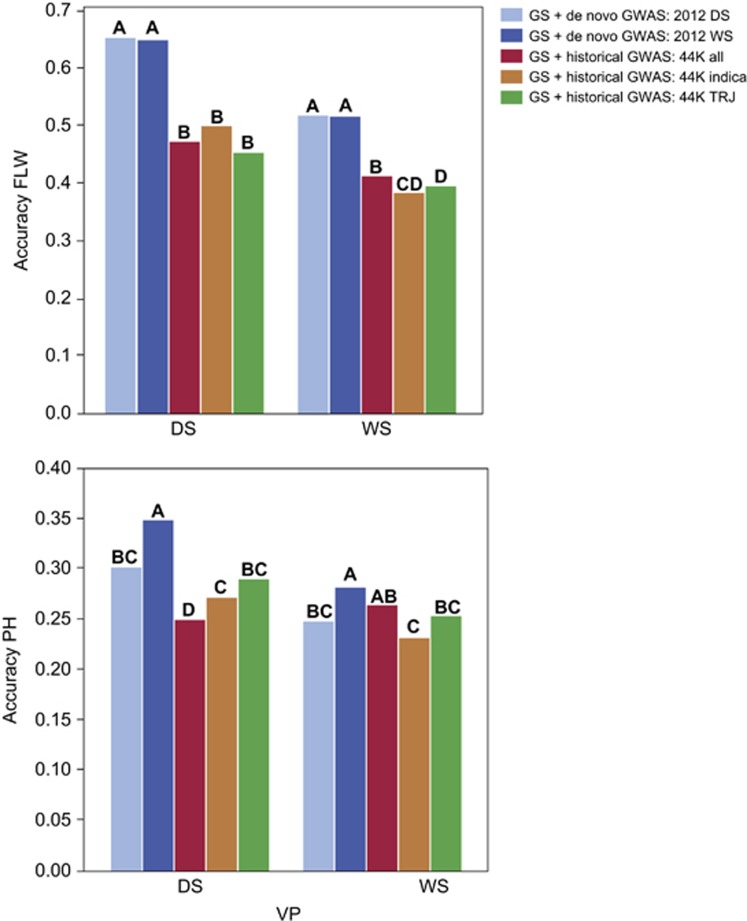
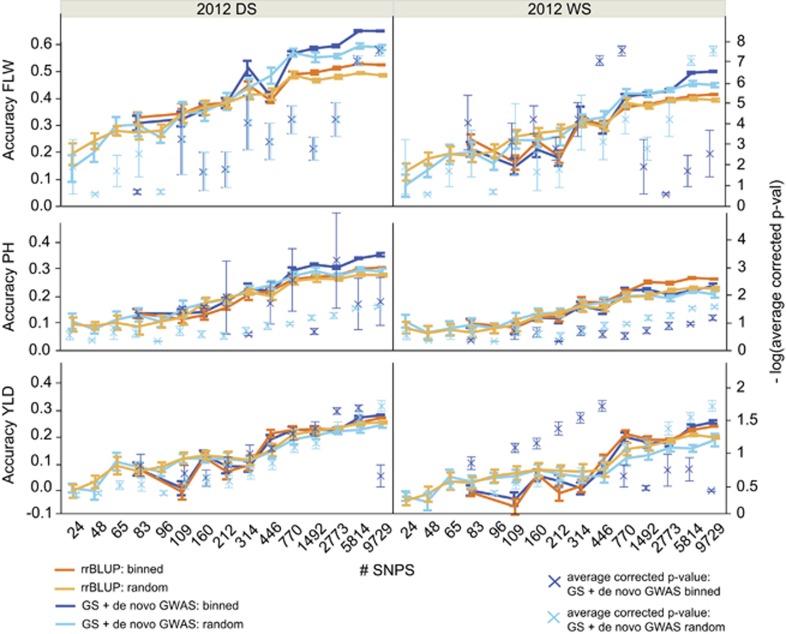
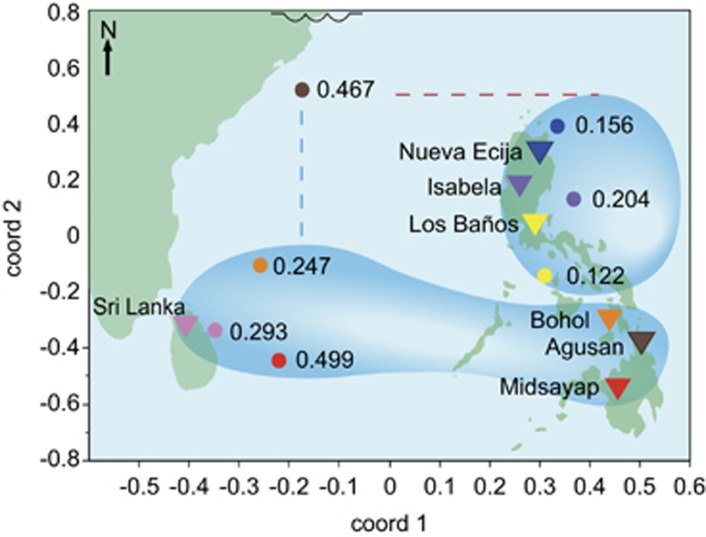
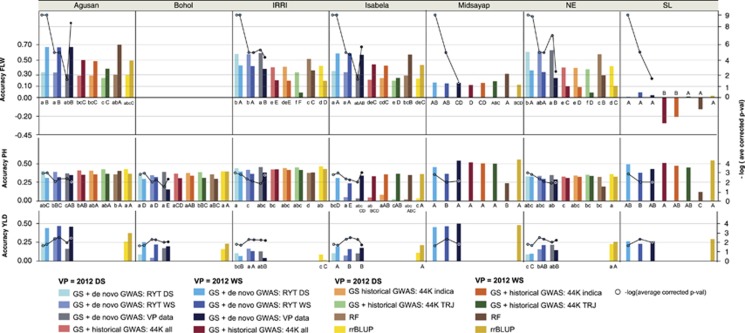
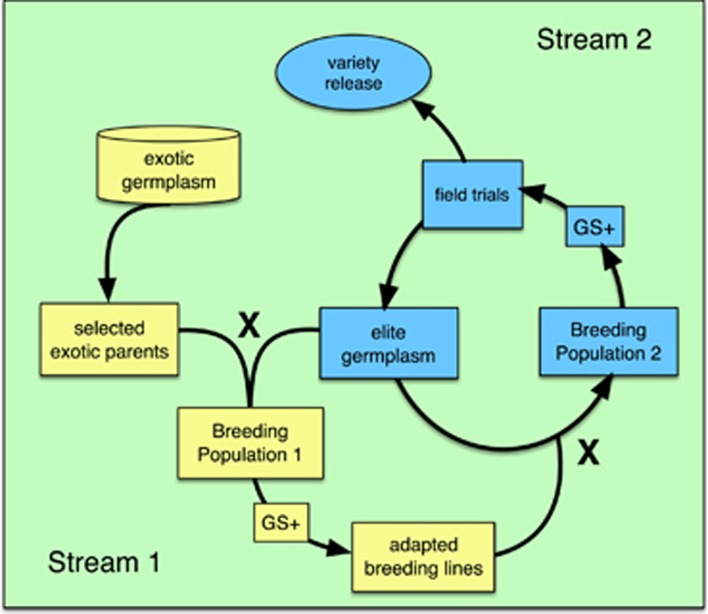
To address the multiple challenges to food security posed by global climate change, population growth and rising incomes, plant breeders are developing new crop varieties that can enhance both agricultural productivity and environmental sustainability. Current breeding practices, however, are unable to keep pace with demand. Genomic selection (GS) is a new technique that helps accelerate the rate of genetic gain in breeding by using whole-genome data to predict the breeding value of offspring. Here, we describe a new GS model that combines RR-BLUP with markers fit as fixed effects selected from the results of a genome-wide-association study (GWAS) on the RR-BLUP training data. We term this model GS + de novo GWAS. In a breeding population of tropical rice, GS + de novo GWAS outperformed six other models for a variety of traits and in multiple environments. On the basis of these results, we propose an extended, two-part breeding design that can be used to efficiently integrate novel variation into elite breeding populations, thus expanding genetic diversity and enhancing the potential for sustainable productivity gains.
Figures
References
-
- Asoro FG, Newell MA, Beavis WD, Scott MP, Tinker NA, Jannink J-L. (2013). Genomic, marker-assisted, and pedigree-BLUP selection methods for β-glucan concentration in elite oat. Crop Sci 53: 1894–1906.
-
- Benjamini Y, Hochberg Y. (1995). Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Statist Soc B 57: 289–300.
-
- Bentley AR, Scutari M, Gosman N, Faure S, Bedford F, Howell P et al. (2014). Applying association mapping and genomic selection to the dissection of key traits in elite European wheat. Theor Appl Genet 127: 2619–2633. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
