

When Quality Beats Quantity: Decision Theory, Drug Discovery, and the Reproducibility Crisis
- PMID: 26863229
- PMCID: PMC4749240
- DOI: 10.1371/journal.pone.0147215
When Quality Beats Quantity: Decision Theory, Drug Discovery, and the Reproducibility Crisis
Abstract
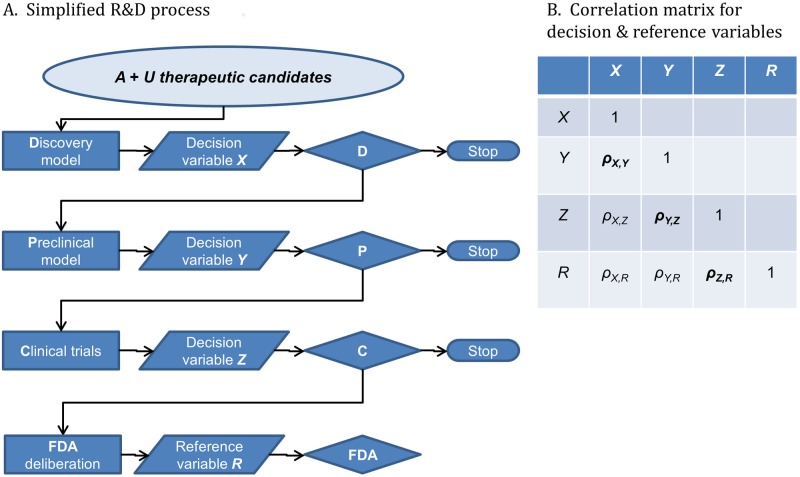
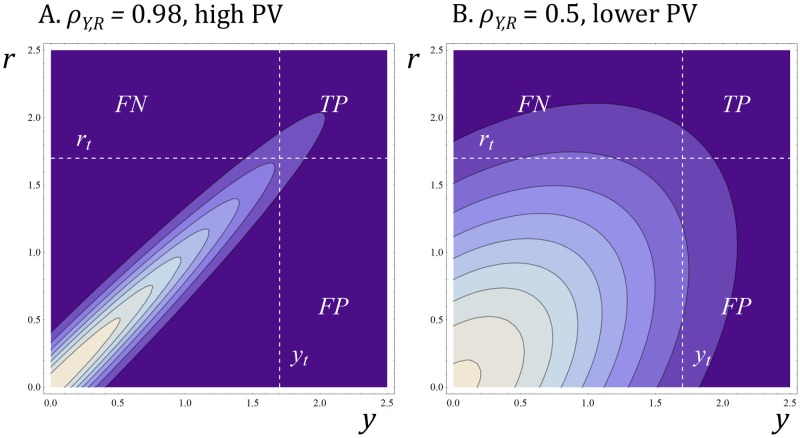
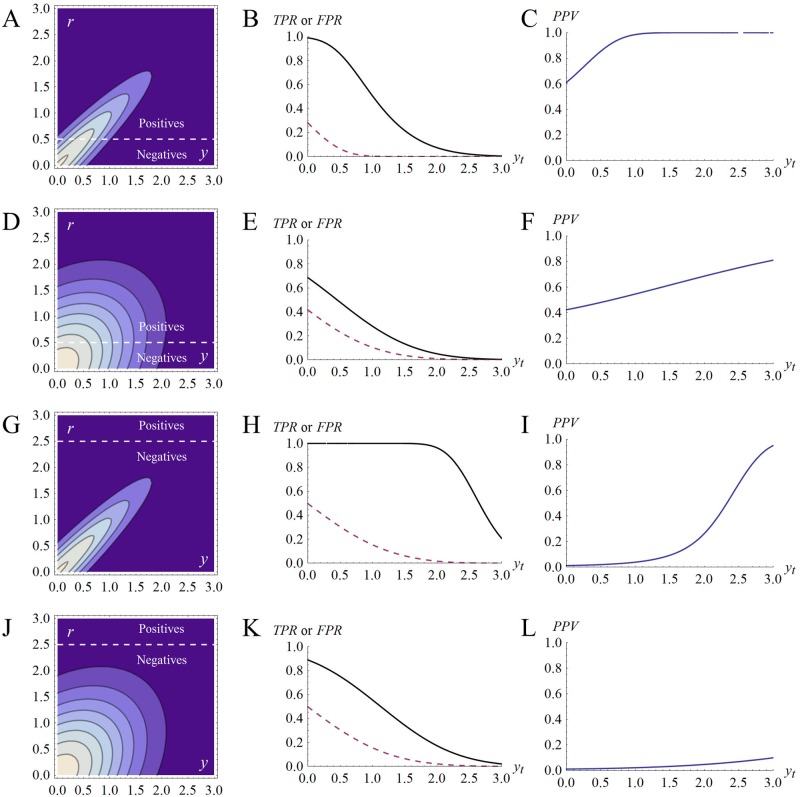
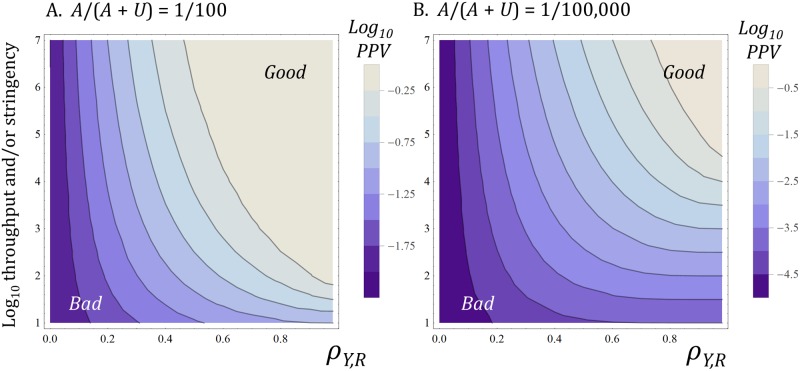
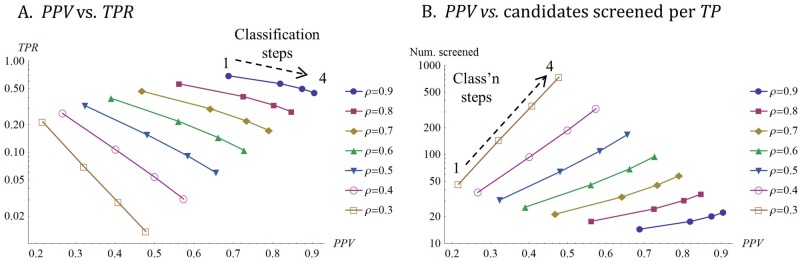
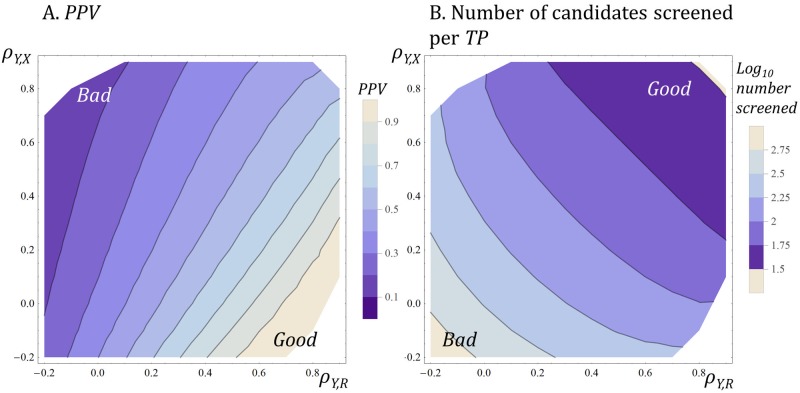
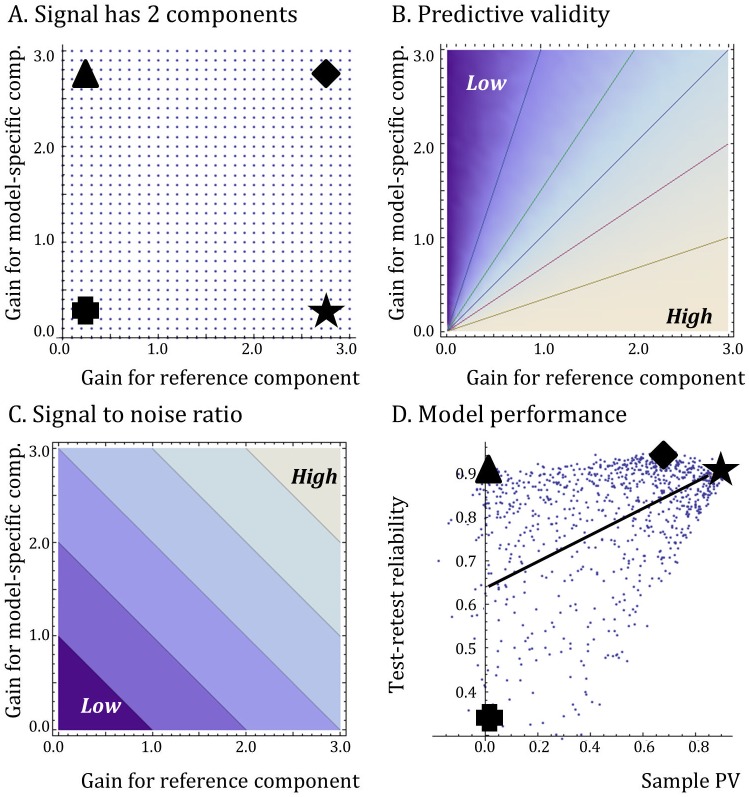
A striking contrast runs through the last 60 years of biopharmaceutical discovery, research, and development. Huge scientific and technological gains should have increased the quality of academic science and raised industrial R&D efficiency. However, academia faces a "reproducibility crisis"; inflation-adjusted industrial R&D costs per novel drug increased nearly 100 fold between 1950 and 2010; and drugs are more likely to fail in clinical development today than in the 1970s. The contrast is explicable only if powerful headwinds reversed the gains and/or if many "gains" have proved illusory. However, discussions of reproducibility and R&D productivity rarely address this point explicitly. The main objectives of the primary research in this paper are: (a) to provide quantitatively and historically plausible explanations of the contrast; and (b) identify factors to which R&D efficiency is sensitive. We present a quantitative decision-theoretic model of the R&D process. The model represents therapeutic candidates (e.g., putative drug targets, molecules in a screening library, etc.) within a "measurement space", with candidates' positions determined by their performance on a variety of assays (e.g., binding affinity, toxicity, in vivo efficacy, etc.) whose results correlate to a greater or lesser degree. We apply decision rules to segment the space, and assess the probability of correct R&D decisions. We find that when searching for rare positives (e.g., candidates that will successfully complete clinical development), changes in the predictive validity of screening and disease models that many people working in drug discovery would regard as small and/or unknowable (i.e., an 0.1 absolute change in correlation coefficient between model output and clinical outcomes in man) can offset large (e.g., 10 fold, even 100 fold) changes in models' brute-force efficiency. We also show how validity and reproducibility correlate across a population of simulated screening and disease models. We hypothesize that screening and disease models with high predictive validity are more likely to yield good answers and good treatments, so tend to render themselves and their diseases academically and commercially redundant. Perhaps there has also been too much enthusiasm for reductionist molecular models which have insufficient predictive validity. Thus we hypothesize that the average predictive validity of the stock of academically and industrially "interesting" screening and disease models has declined over time, with even small falls able to offset large gains in scientific knowledge and brute-force efficiency. The rate of creation of valid screening and disease models may be the major constraint on R&D productivity.
Conflict of interest statement
Figures
References
-
- Hogan JC. Combinatorial chemistry in drug discovery. Nat Biotechnol. 1997; 15: p. 328–330. - PubMed
-
- Geysen HM, Schoenen F, Wagner D, Wagner R. Combinatorial compound libraries for drug discovery: an ongoing challenge. Nat Rev Drug Discov. 2003; 2: p. 222–230. - PubMed
-
- Nature Biotechnology. Combinatorial chemistry. Nat Biotechnol. 2000; 18 supplement: p. IT50–IT52. - PubMed
-
- Dolle RE. Historical overview of chemical library design In Zhou JZ, editor. Chemical Library Design (Methods in Molecular Biology 685).: Springer Science; 2011. p. 3–25. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
