

Integrative modeling of multi-omics data to identify cancer drivers and infer patient-specific gene activity
- PMID: 26864072
- PMCID: PMC4750289
- DOI: 10.1186/s12918-016-0260-9
Integrative modeling of multi-omics data to identify cancer drivers and infer patient-specific gene activity
Abstract
Background: High throughput technologies have been used to profile genes in multiple different dimensions, such as genetic variation, copy number, gene and protein expression, epigenetics, metabolomics. Computational analyses often treat these different data types as independent, leading to an explosion in the number of features making studies under-powered and more importantly do not provide a comprehensive view of the gene's state. We sought to infer gene activity by integrating different dimensions using biological knowledge of oncogenes and tumor suppressors.
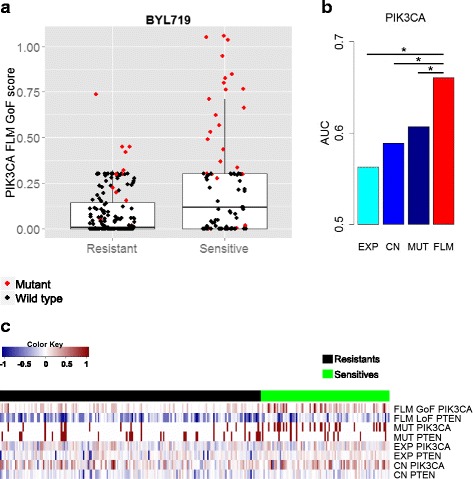
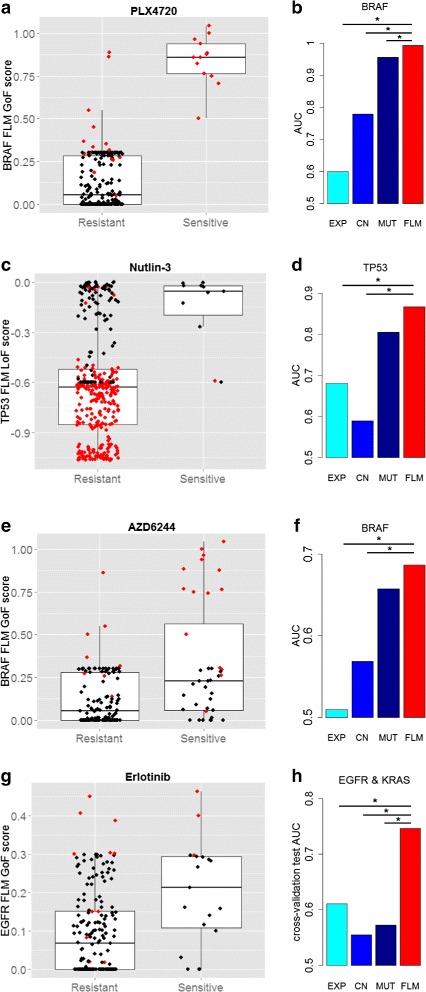
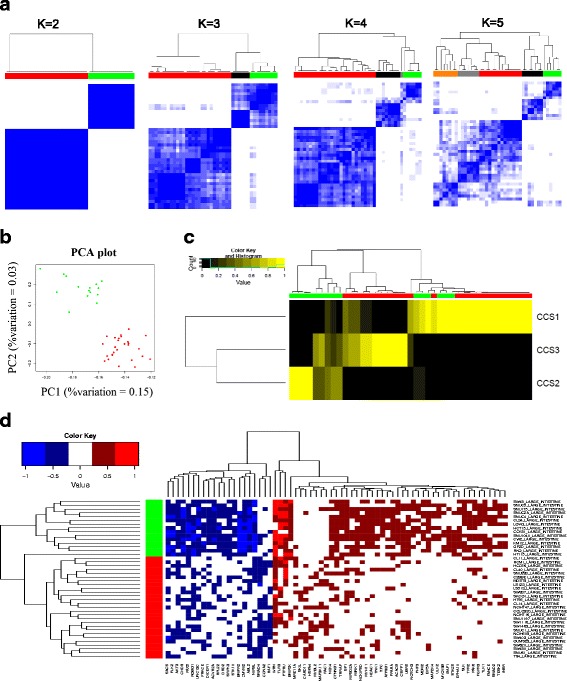
Results: This paper proposes an integrative model of oncogene and tumor suppressor activity in cells which is used to identify cancer drivers and compute patient-specific gene activity scores. We have developed a Fuzzy Logic Modeling (FLM) framework to incorporate biological knowledge with multi-omics data such as somatic mutation, gene expression and copy number measurements. The advantage of using a fuzzy logic approach is to abstract meaningful biological rules from low-level numerical data. Biological knowledge is often qualitative, thus combining it with quantitative numerical measurements may leverage new biological insights about a gene's state. We show that the oncogenic and altered tumor suppressing state of a gene can be better characterized by integrating different molecular measurements with biological knowledge than by each data type alone. We validate the gene activity score using data from the Cancer Cell Line Encyclopedia and drug sensitivity data for five compounds: BYL719 (PIK3CA inhibitor), PLX4720 (BRAF inhibitor), AZD6244 (MEK inhibitor), Erlotinib (EGFR inhibitor), and Nutlin-3 (MDM2 inhibitor). The integrative score improves prediction of drug sensitivity for the known drug targets of these compounds compared to each data type alone. The gene activity scores are also used to cluster colorectal cancer cell lines. Two subtypes of CRCs were found and potential cancer drivers and therapeutic targets for each of the subtypes were identified.
Conclusions: We propose a fuzzy logic based approach to infer gene activity in cancer by integrating numerical data with descriptive biological knowledge. We compute general patient-specific gene-level scores useful to determine the oncogenic or tumor suppressor status of cancer gene drivers and to cluster or classify patients.
Figures


References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
