

Digital Sorting of Pure Cell Populations Enables Unambiguous Genetic Analysis of Heterogeneous Formalin-Fixed Paraffin-Embedded Tumors by Next Generation Sequencing
- PMID: 26864208
- PMCID: PMC4750064
- DOI: 10.1038/srep20944
Digital Sorting of Pure Cell Populations Enables Unambiguous Genetic Analysis of Heterogeneous Formalin-Fixed Paraffin-Embedded Tumors by Next Generation Sequencing
Abstract
Precision medicine in oncology requires an accurate characterization of a tumor molecular profile for patient stratification. Though targeted deep sequencing is an effective tool to detect the presence of somatic sequence variants, a significant number of patient specimens do not meet the requirements needed for routine clinical application. Analysis is hindered by contamination of normal cells and inherent tumor heterogeneity, compounded with challenges of dealing with minute amounts of tissue and DNA damages common in formalin-fixed paraffin-embedded (FFPE) specimens. Here we present an innovative workflow using DEPArray™ system, a microchip-based digital sorter to achieve 100%-pure, homogenous subpopulations of cells from FFPE samples. Cells are distinguished by fluorescently labeled antibodies and DNA content. The ability to address tumor heterogeneity enables unambiguous determination of true-positive sequence variants, loss-of-heterozygosity as well as copy number variants. The proposed strategy overcomes the inherent trade-offs made between sensitivity and specificity in detecting genetic variants from a mixed population, thus rescuing for analysis even the smaller clinical samples with low tumor cellularity.
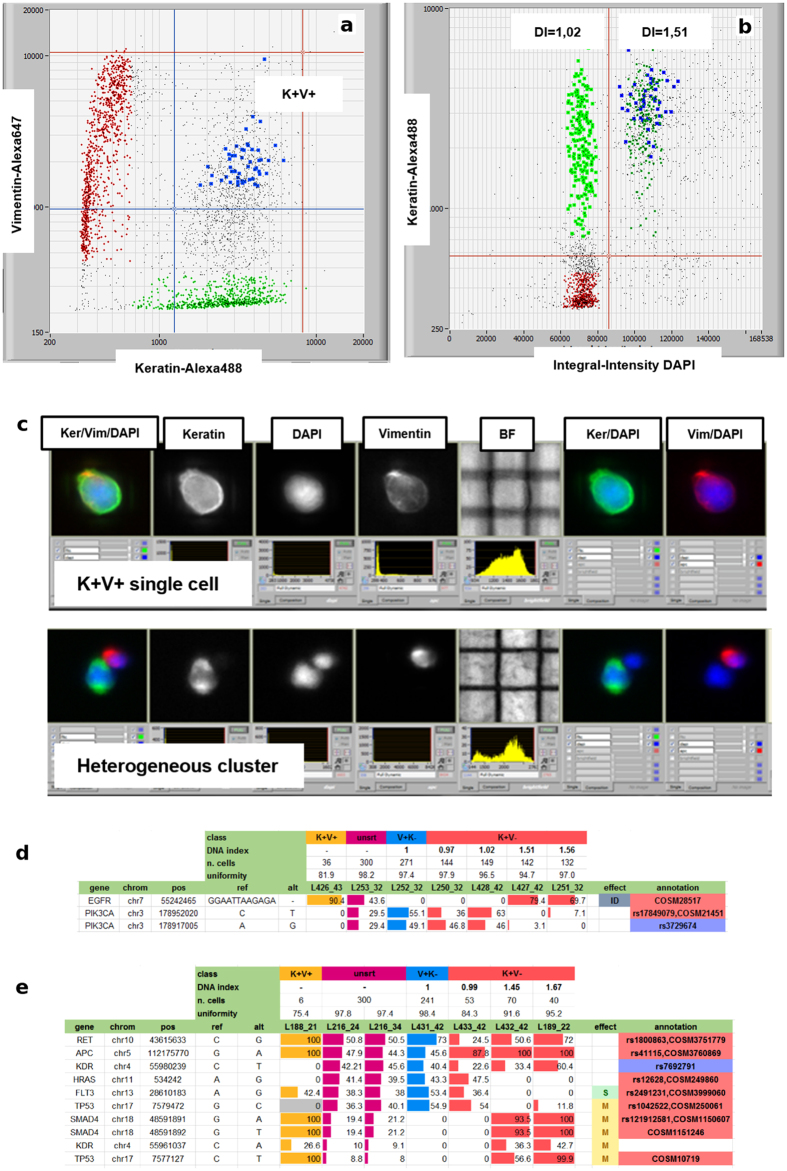
Figures





References
-
- Do H. & Dobrovic A. Sequence artifacts in DNA from formalin-fixed tissues: causes and strategies for minimization. Clin. Chem. 61, 64–71 (2015). - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
