

Explaining the geographic spread of emerging epidemics: a framework for comparing viral phylogenies and environmental landscape data
- PMID: 26864798
- PMCID: PMC4750353
- DOI: 10.1186/s12859-016-0924-x
Explaining the geographic spread of emerging epidemics: a framework for comparing viral phylogenies and environmental landscape data
Abstract
Background: Phylogenetic analysis is now an important tool in the study of viral outbreaks. It can reconstruct epidemic history when surveillance epidemiology data are sparse, and can indicate transmission linkages among infections that may not otherwise be evident. However, a remaining challenge is to develop an analytical framework that can test hypotheses about the effect of environmental variables on pathogen spatial spread. Recent phylogeographic approaches can reconstruct the history of virus dispersal from sampled viral genomes and infer the locations of ancestral infections. Such methods provide a unique source of spatio-temporal information, and are exploited here.
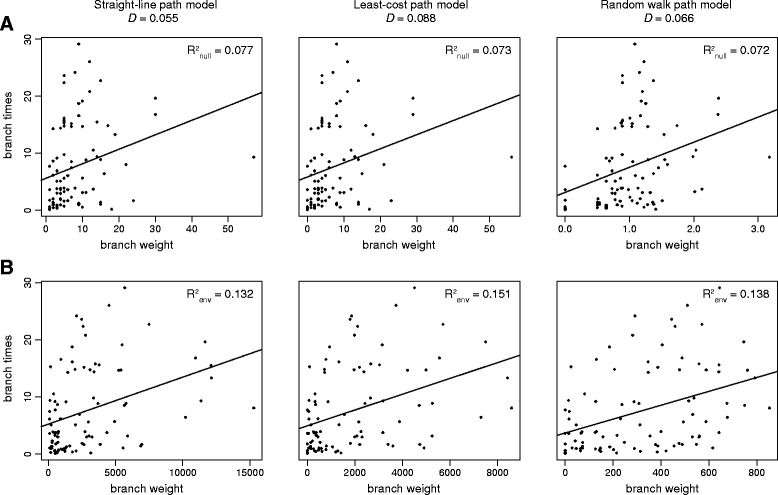
Results: We present and apply a new statistical framework that combines genomic and geographic data to test the impact of environmental variables on the mode and tempo of pathogen dispersal during emerging epidemics. First, the spatial history of an emerging pathogen is estimated using standard phylogeographic methods. The inferred dispersal path for each phylogenetic lineage is then assigned a "weight" using environmental data (e.g. altitude, land cover). Next, tests measure the association between each environmental variable and lineage movement. A randomisation procedure is used to assess statistical confidence and we validate this approach using simulated data. We apply our new framework to a set of gene sequences from an epidemic of rabies virus in North American raccoons. We test the impact of six different environmental variables on this epidemic and demonstrate that elevation is associated with a slower rabies spread in a natural population.
Conclusion: This study shows that it is possible to integrate genomic and environmental data in order to test hypotheses concerning the mode and tempo of virus dispersal during emerging epidemics.
Figures




References
-
- Corman VM, Ithete NL, Richards LR, Schoeman MC, Preiser W, Drosten C, et al. Rooting the phylogenetic tree of Middle East respiratory syndrome coronavirus by characterization of a conspecific virus from an African bat. J Virol. 2014;88(19):11297–11303. doi: 10.1128/JVI.01498-14. - DOI - PMC - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
