

Use of whole-genome sequencing to trace, control and characterize the regional expansion of extended-spectrum β-lactamase producing ST15 Klebsiella pneumoniae
- PMID: 26864946
- PMCID: PMC4749987
- DOI: 10.1038/srep20840
Use of whole-genome sequencing to trace, control and characterize the regional expansion of extended-spectrum β-lactamase producing ST15 Klebsiella pneumoniae
Abstract
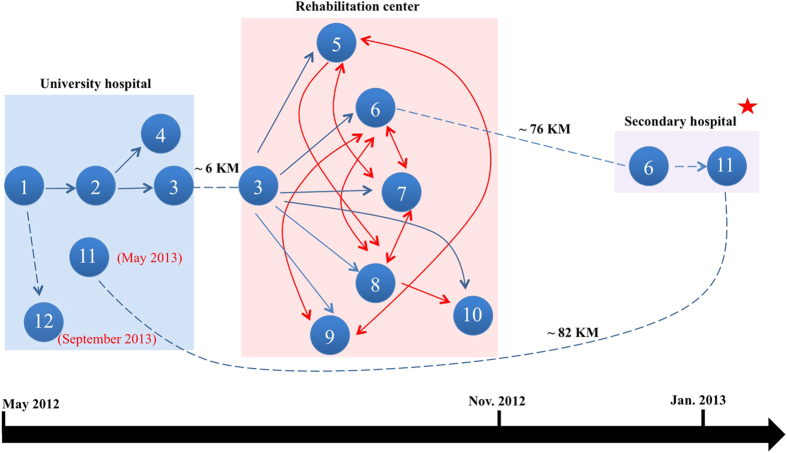
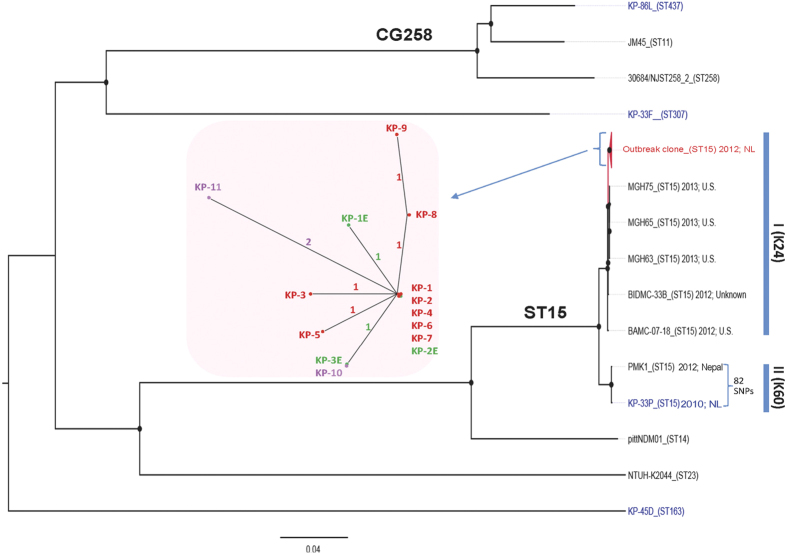
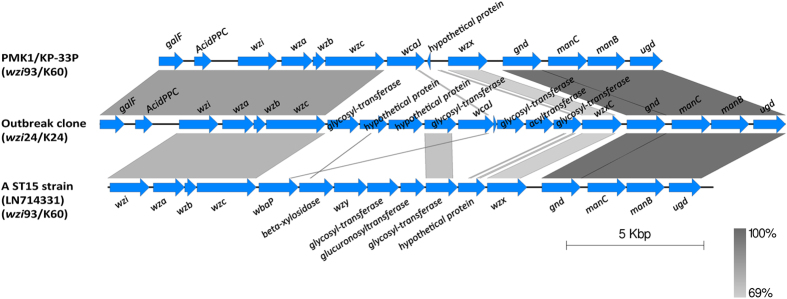
The study describes the transmission of a CTX-M-15-producing ST15 Klebsiella pneumoniae between patients treated in a single center and the subsequent inter-institutional spread by patient referral occurring between May 2012 and September 2013. A suspected epidemiological link between clinical K. pneumoniae isolates was supported by patient contact tracing and genomic phylogenetic analysis from May to November 2012. By May 2013, a patient treated in three institutions in two cities was involved in an expanding cluster caused by this high-risk clone (HiRiC) (local expansion, CTX-M-15 producing, and containing hypervirulence factors). A clone-specific multiplex PCR was developed for patient screening by which another patient was identified in September 2013. Genomic phylogenetic analysis including published ST15 genomes revealed a close homology with isolates previously found in the USA. Environmental contamination and lack of consistent patient screening were identified as being responsible for the clone dissemination. The investigation addresses the advantages of whole-genome sequencing in the early detection of HiRiC with a high propensity of nosocomial transmission and prolonged circulation in the regional patient population. Our study suggests the necessity for inter-institutional/regional collaboration for infection/outbreak management of K. pneumoniae HiRiCs.
Figures
Similar articles
-
Expansion and countrywide dissemination of ST11, ST15 and ST147 ciprofloxacin-resistant CTX-M-15-type beta-lactamase-producing Klebsiella pneumoniae epidemic clones in Hungary in 2005--the new 'MRSAs'?J Antimicrob Chemother. 2008 Nov;62(5):978-85. doi: 10.1093/jac/dkn287. Epub 2008 Jul 30. J Antimicrob Chemother. 2008. PMID: 18667450
-
Hospital cross-transmission of extended-spectrum β-lactamase producing Escherichia coli and Klebsiella pneumoniae.Med Mal Infect. 2013 Aug;43(8):331-6. doi: 10.1016/j.medmal.2013.06.001. Epub 2013 Jul 19. Med Mal Infect. 2013. PMID: 23876202
-
High clonal diversity in a non-outbreak situation of clinical ESBL-producing Klebsiella pneumoniae isolates in the first national surveillance program in Cuba.Microb Drug Resist. 2014 Feb;20(1):45-51. doi: 10.1089/mdr.2013.0021. Epub 2013 May 21. Microb Drug Resist. 2014. PMID: 23692050
-
Identification and characterization of CTX-M-15 producing Klebsiella pneumoniae clone ST101 in a Hungarian university teaching hospital.Acta Microbiol Immunol Hung. 2015 Sep;62(3):233-45. doi: 10.1556/030.62.2015.3.2. Acta Microbiol Immunol Hung. 2015. PMID: 26551567 Review.
-
Carbapenemase-Producing Klebsiella pneumoniae, a Key Pathogen Set for Global Nosocomial Dominance.Antimicrob Agents Chemother. 2015 Oct;59(10):5873-84. doi: 10.1128/AAC.01019-15. Epub 2015 Jul 13. Antimicrob Agents Chemother. 2015. PMID: 26169401 Free PMC article. Review.
Cited by
-
A Front Line on Klebsiella pneumoniae Capsular Polysaccharide Knowledge: Fourier Transform Infrared Spectroscopy as an Accurate and Fast Typing Tool.mSystems. 2020 Mar 24;5(2):e00386-19. doi: 10.1128/mSystems.00386-19. mSystems. 2020. PMID: 32209717 Free PMC article.
-
A mathematical model and inference method for bacterial colonization in hospital units applied to active surveillance data for carbapenem-resistant enterobacteriaceae.PLoS One. 2020 Nov 12;15(11):e0231754. doi: 10.1371/journal.pone.0231754. eCollection 2020. PLoS One. 2020. PMID: 33180781 Free PMC article.
-
Clinical and Molecular Description of a High-Copy IncQ1 KPC-2 Plasmid Harbored by the International ST15 Klebsiella pneumoniae Clone.mSphere. 2020 Oct 7;5(5):e00756-20. doi: 10.1128/mSphere.00756-20. mSphere. 2020. PMID: 33028683 Free PMC article.
-
Surveillance of carbapenem-resistant organisms using next-generation sequencing.Front Public Health. 2023 May 15;11:1184045. doi: 10.3389/fpubh.2023.1184045. eCollection 2023. Front Public Health. 2023. PMID: 37255756 Free PMC article.
-
HAIviz: an interactive dashboard for visualising and integrating healthcare-associated genomic epidemiological data.Microb Genom. 2024 Feb;10(2):001200. doi: 10.1099/mgen.0.001200. Microb Genom. 2024. PMID: 38358326 Free PMC article.
References
-
- Breurec S. et al.. Klebsiella pneumoniae resistant to third-generation cephalosporins in five African and two Vietnamese major towns: multiclonal population structure with two major international clonal groups, CG15 and CG258. Clin. Microbiol. Infect. 19, 349–355, doi: 10.1111/j.1469-0691.2012.03805.x (2013). - DOI - PubMed
-
- Damjanova I. et al.. Expansion and countrywide dissemination of ST11, ST15 and ST147 ciprofloxacin-resistant CTX-M-15-type β-lactamase-producing Klebsiella pneumoniae epidemic clones in Hungary in 2005— the new ‘MRSAs’? J. Antimicrob. Chemother. 62, 978–985, doi: 10.1093/jac/dkn287 (2008). - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
