

Altered Trafficking and Processing of GALC Mutants Correlates with Globoid Cell Leukodystrophy Severity
- PMID: 26865610
- PMCID: PMC4748072
- DOI: 10.1523/JNEUROSCI.3095-15.2016
Altered Trafficking and Processing of GALC Mutants Correlates with Globoid Cell Leukodystrophy Severity
Abstract
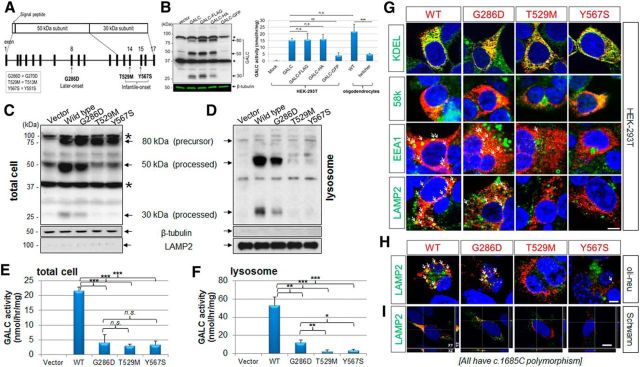
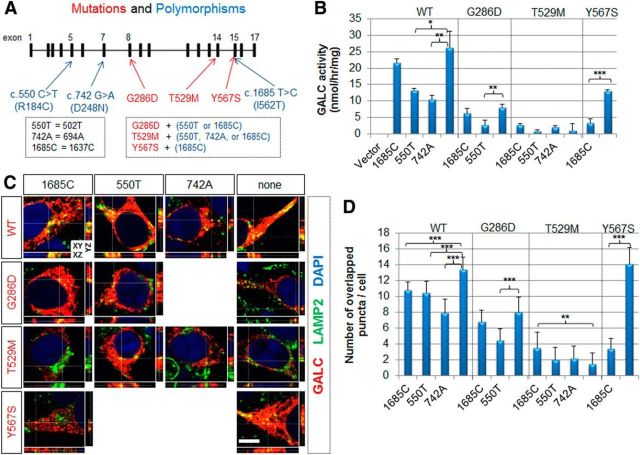
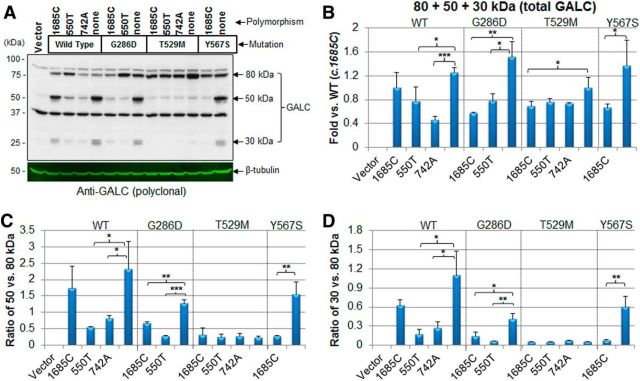
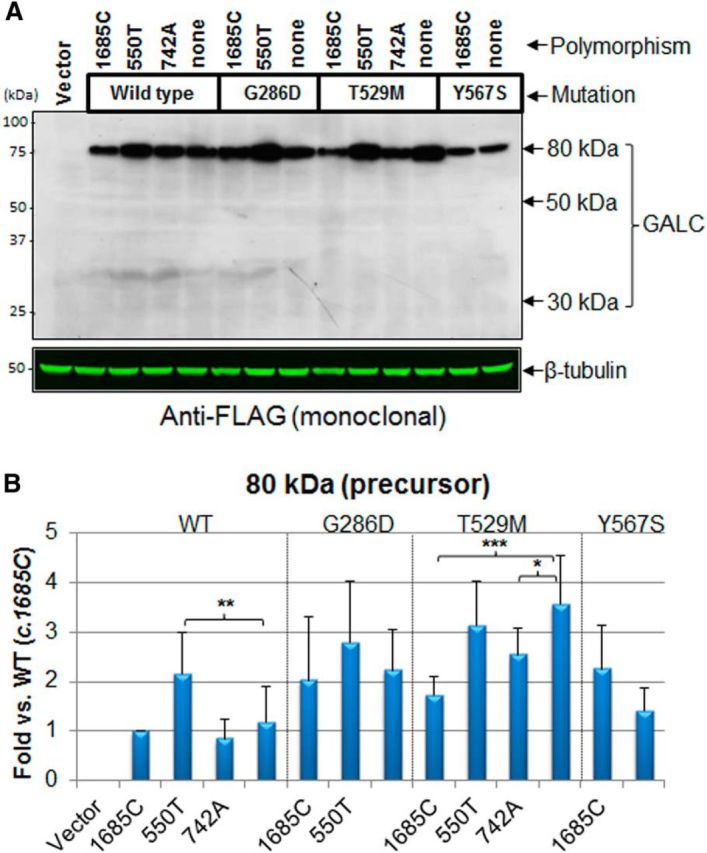
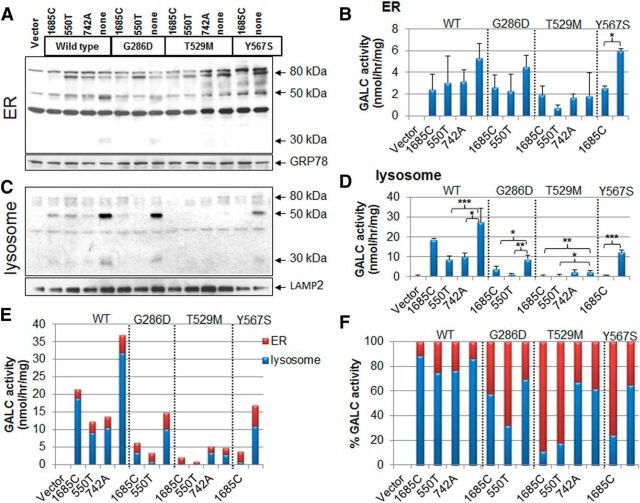
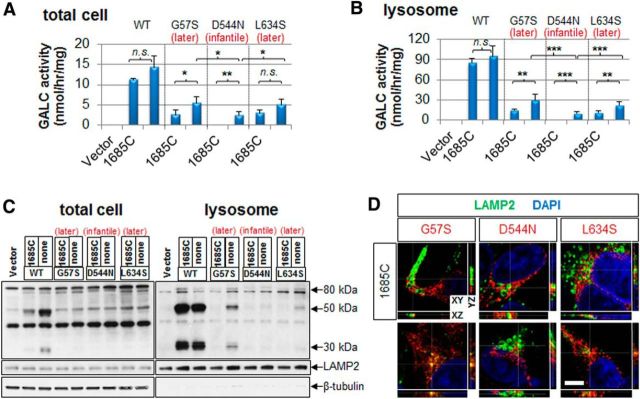
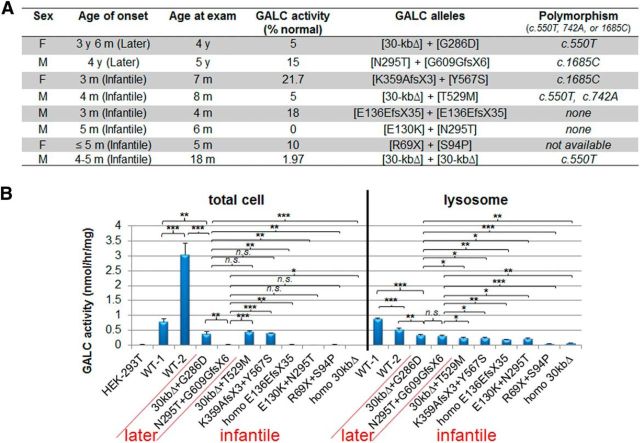
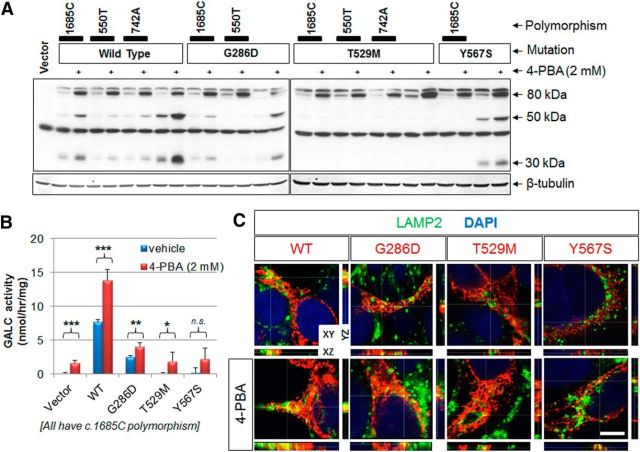
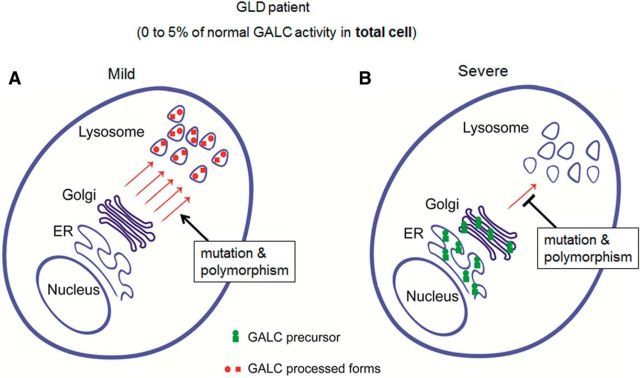
Globoid cell leukodystrophy (GLD, Krabbe disease) is due to autosomal recessive mutations in the lysosomal enzyme galactosylceramidase (GALC). Many GLD patients develop infantile-onset of progressive neurologic deterioration and death by 2 years of age, whereas others have a later-onset, milder disease. Cord blood transplant slows disease progression much more effectively when performed presymptomatically, highlighting the importance of early diagnosis. Current diagnosis is based on reduced GALC activity, DNA sequence, and clinical examination. However, presymptomatic diagnosis is hampered by imperfect genotype-GALC activity-phenotype correlations. In addition, three polymorphisms in the GALC gene are variably associated with disease mutations and have unknown effects on GALC activity and disease outcome. Here, we study mutations that cause infantile or later-onset GLD, and show that GALC activity is significantly lower in infantile versus later-onset mutants when measured in the lysosomal fraction, but not in whole-cell lysates. In parallel, infantile-onset mutant GALCs showed reduced trafficking to lysosomes and processing than later-onset mutant GALCs. Finally, the cis-polymorphisms also affected trafficking to the lysosome and processing of GALC. These differences potentially explain why the activity of different mutations appears similar in whole-cell extracts from lymphocytes, and suggest that measure of GALC activity in lysosomes may better predict the onset and severity of disease for a given GLD genotype.
Significance statement: Globoid cell leukodystrophy (GLD, Krabbe disease) is diagnosed by measuring galactosylceramidase (GALC) activity and DNA analysis. However, genotype and phenotype often do not correlate due to considerable clinical variability, even for the same mutation, for unknown reasons. We find that altered trafficking to the lysosome and processing of GALC correlates with GLD severity and is modulated by cis-polymorphisms. Current diagnosis of GLD is based on GALC activity of total cell lysates from blood, which does not discriminate whether the activity comes from the lysosome or other subcellular organelles. Measurement of GALC activity in lysosomes may predict which infants are at high risk for the infantile phenotype while distinguishing other children who will develop later-onset phenotypes without onset of symptoms for years.
Keywords: Krabbe disease; galactosylceramidase; globoid cell leukodystrophy; lysosomal processing; lysosomal storage disorders; protein trafficking.
Copyright © 2016 the authors 0270-6474/16/361858-13$15.00/0.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical