

Practical Approaches for Detecting Selection in Microbial Genomes
- PMID: 26867134
- PMCID: PMC4750996
- DOI: 10.1371/journal.pcbi.1004739
Practical Approaches for Detecting Selection in Microbial Genomes
Abstract
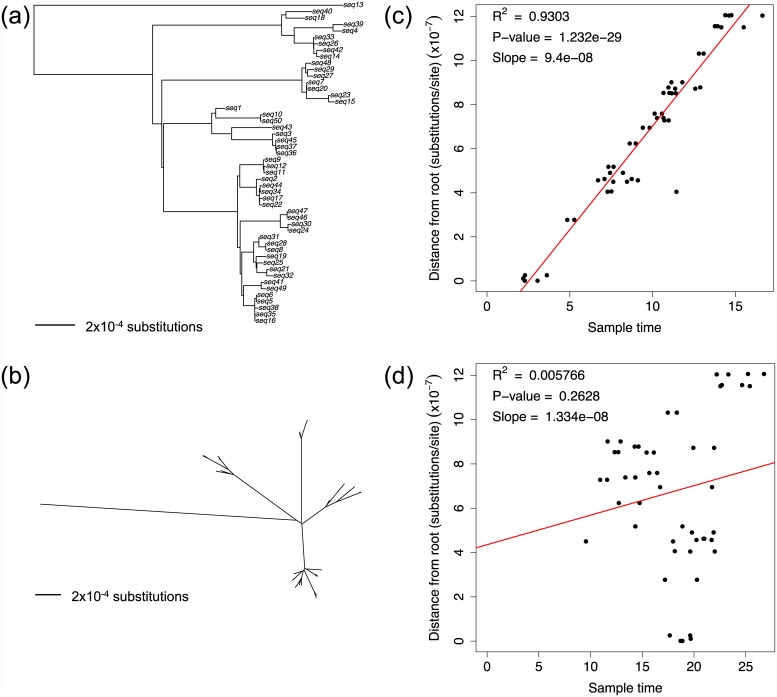
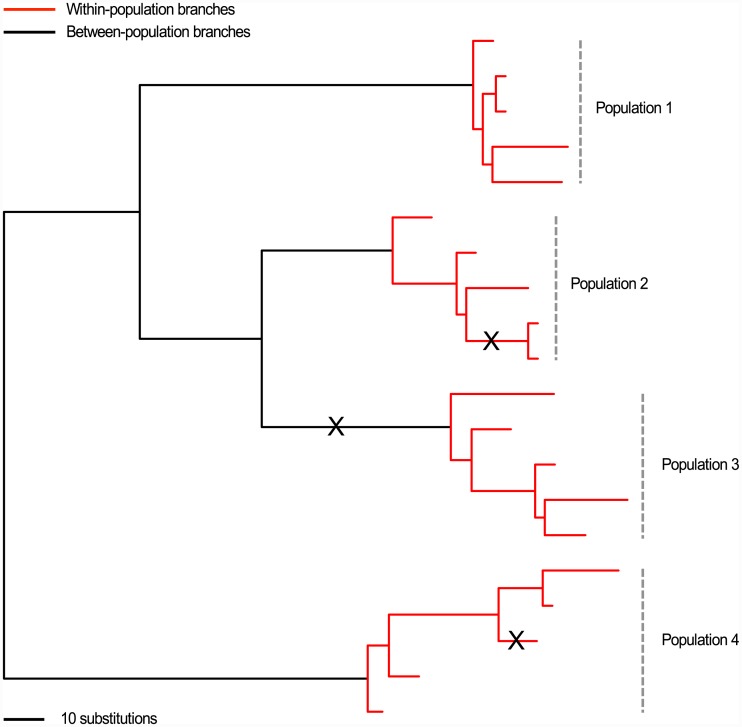
Microbial genome evolution is shaped by a variety of selective pressures. Understanding how these processes occur can help to address important problems in microbiology by explaining observed differences in phenotypes, including virulence and resistance to antibiotics. Greater access to whole-genome sequencing provides microbiologists with the opportunity to perform large-scale analyses of selection in novel settings, such as within individual hosts. This tutorial aims to guide researchers through the fundamentals underpinning popular methods for measuring selection in pathogens. These methods are transferable to a wide variety of organisms, and the exercises provided are designed for researchers with any level of programming experience.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
