

Prostaglandin I₂ Attenuates Prostaglandin E₂-Stimulated Expression of Interferon γ in a β-Amyloid Protein- and NF-κB-Dependent Mechanism
- PMID: 26869183
- PMCID: PMC4751455
- DOI: 10.1038/srep20879
Prostaglandin I₂ Attenuates Prostaglandin E₂-Stimulated Expression of Interferon γ in a β-Amyloid Protein- and NF-κB-Dependent Mechanism
Erratum in
-
Addendum: Prostaglandin I2 Attenuates Prostaglandin E2-Stimulated Expression of Interferon γ in a β-Amyloid Protein- and NF-κB-Dependent Mechanism.Sci Rep. 2022 May 19;12(1):8394. doi: 10.1038/s41598-022-12462-4. Sci Rep. 2022. PMID: 35589946 Free PMC article. No abstract available.
Abstract
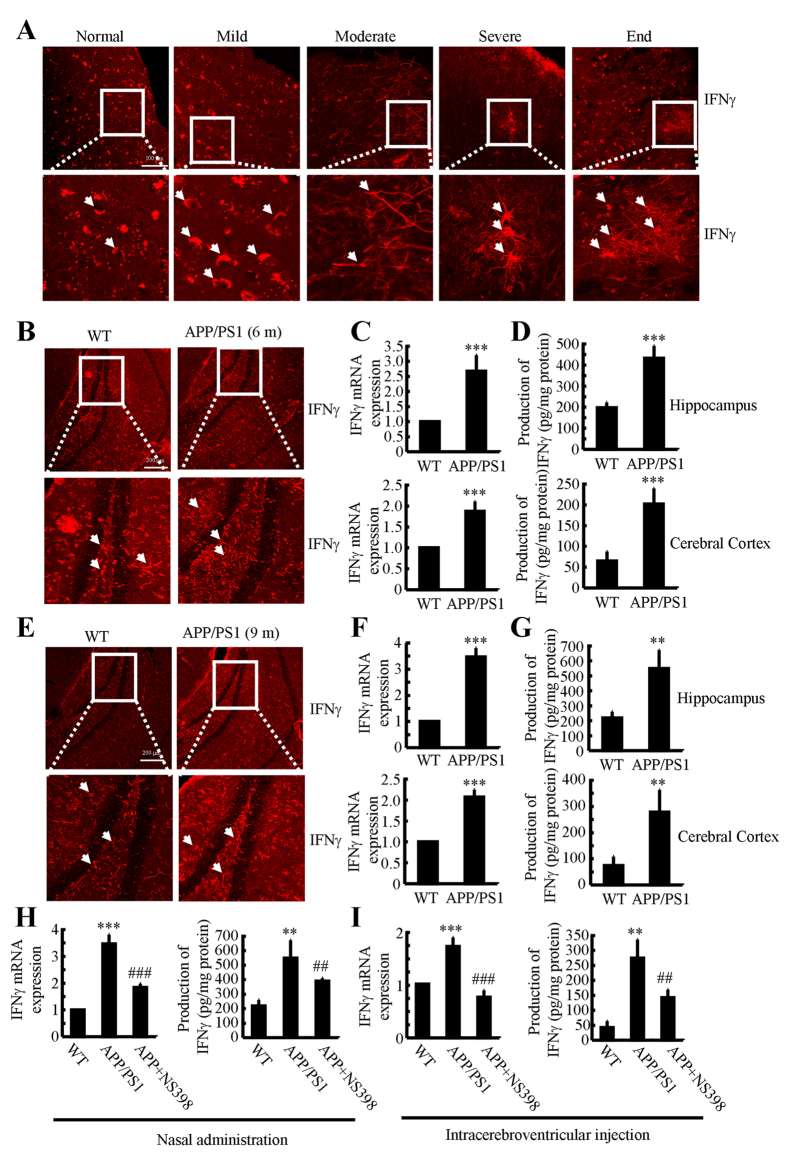
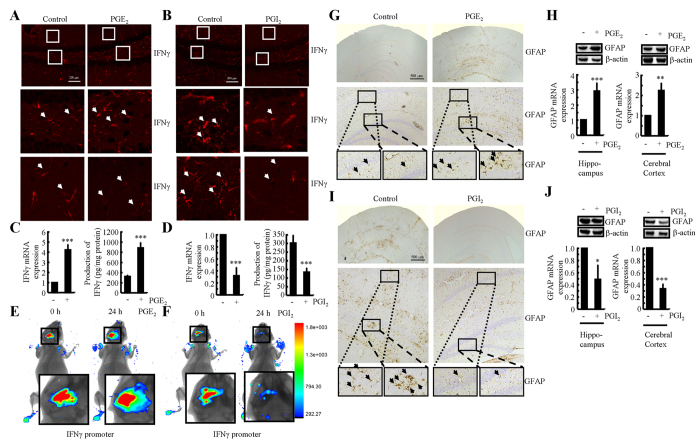
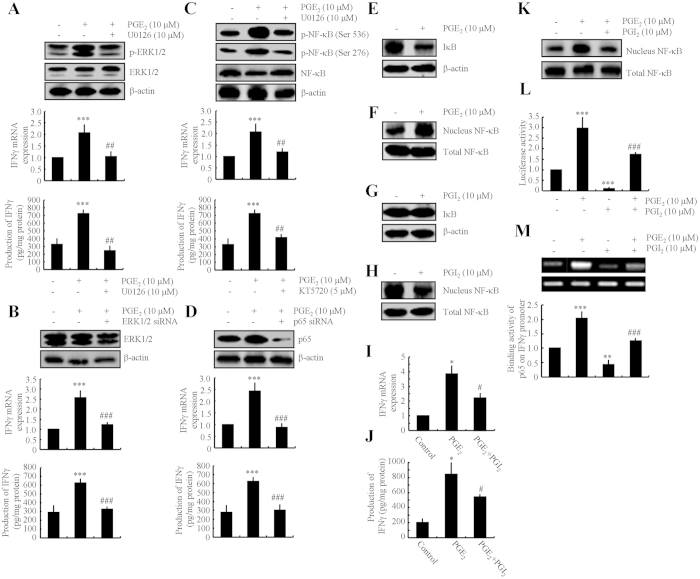
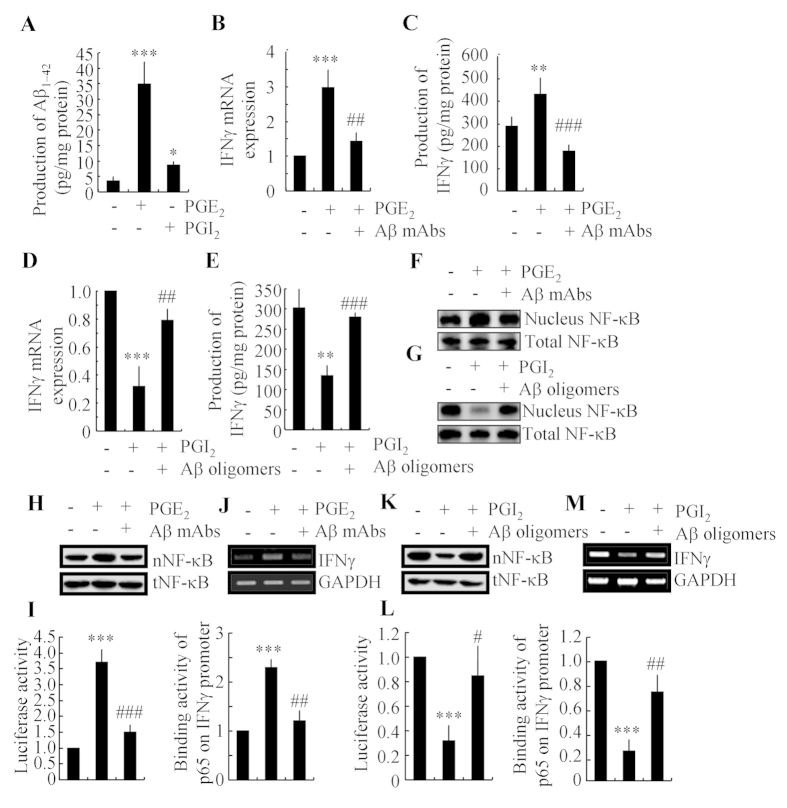
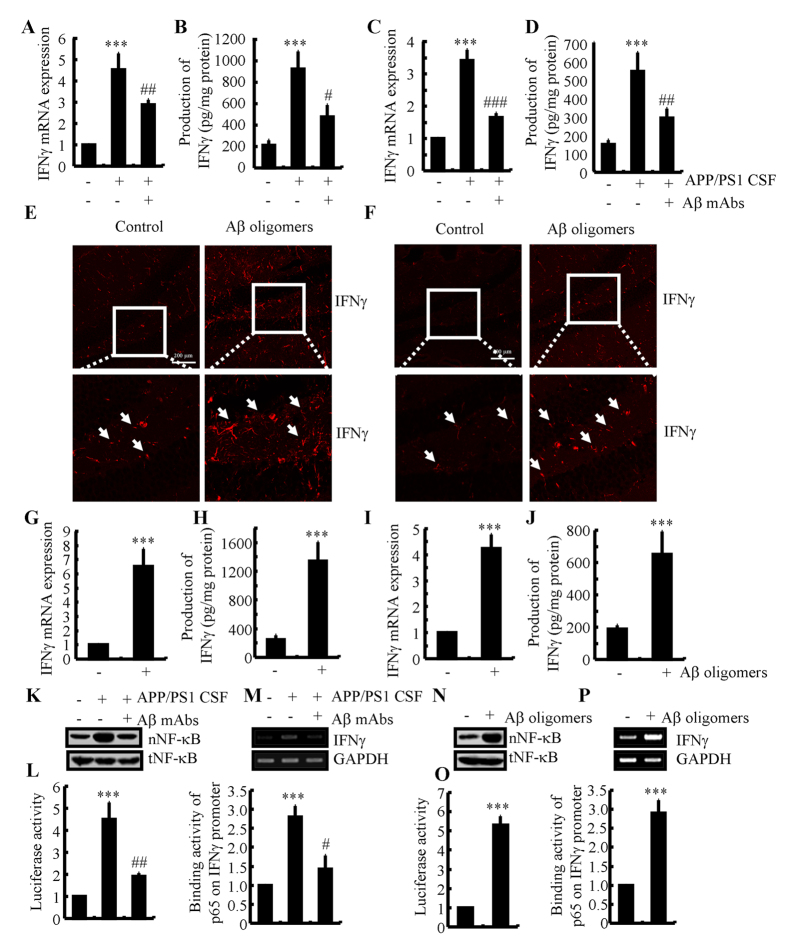
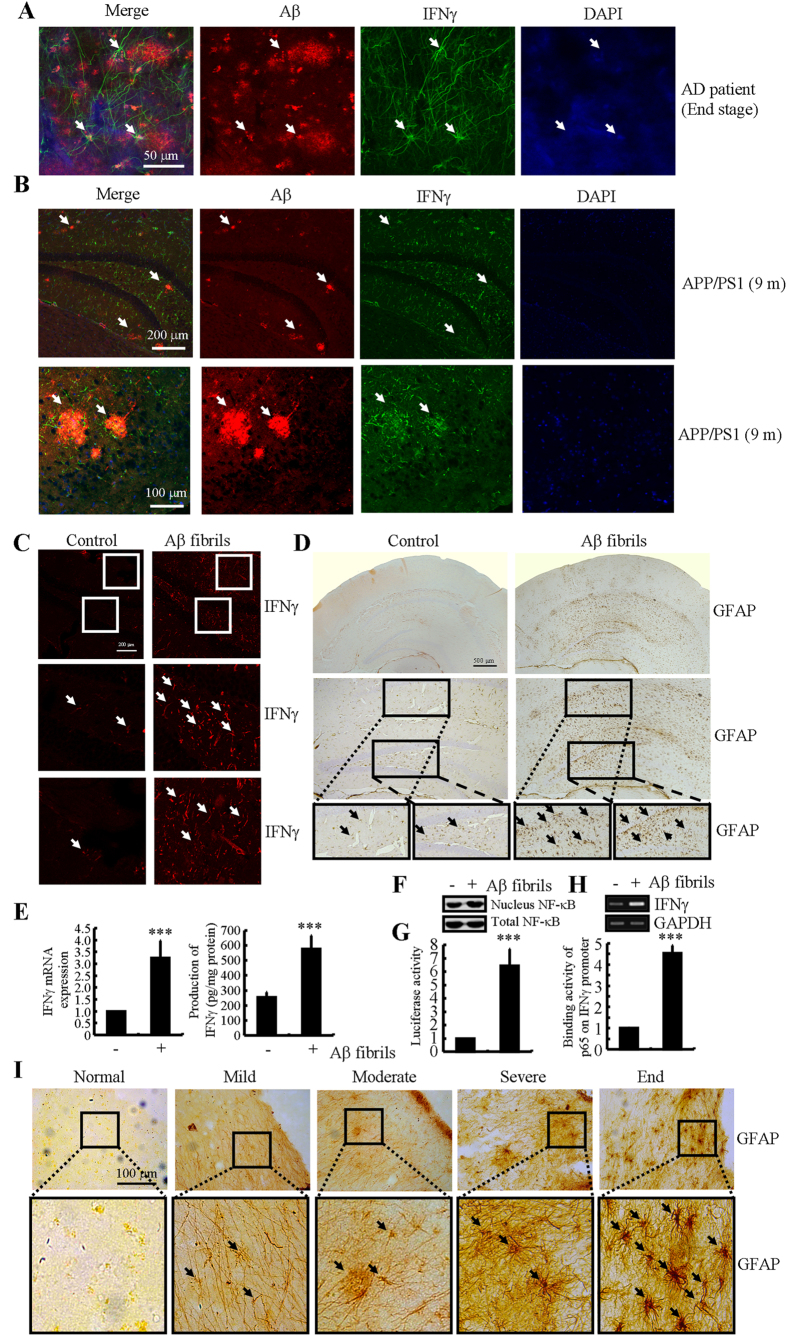
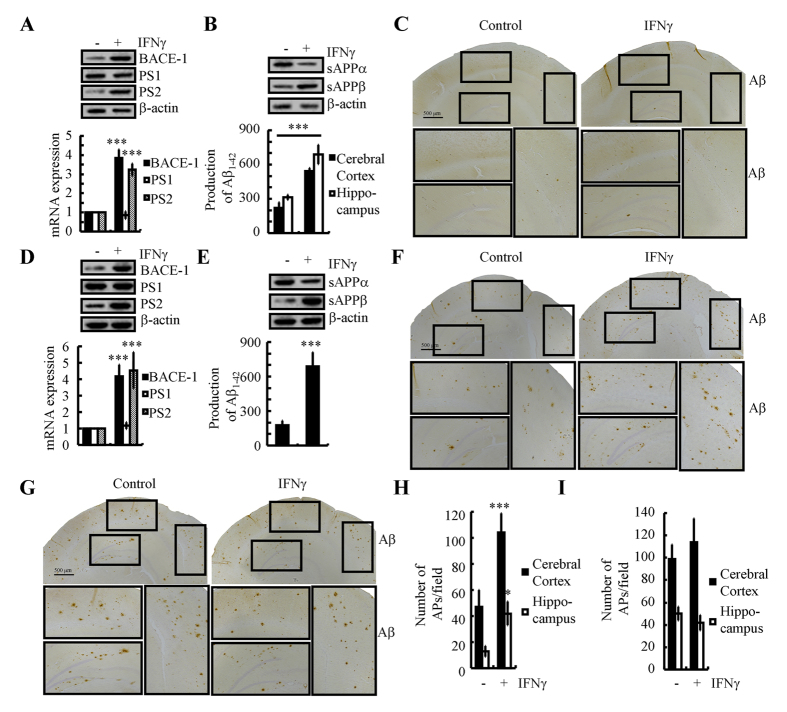
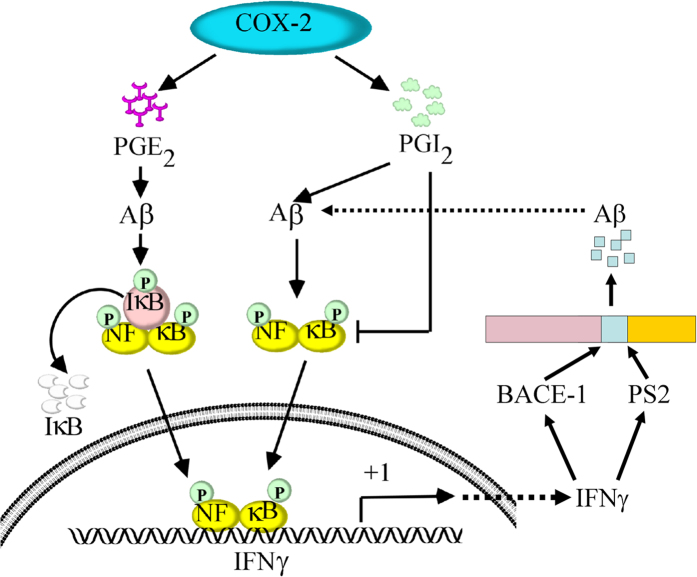
Cyclooxygenase-2 (COX-2) has been recently identified as being involved in the pathogenesis of Alzheimer's disease (AD). However, the role of an important COX-2 metabolic product, prostaglandin (PG) I2, in AD development remains unknown. Using mouse-derived astrocytes as well as APP/PS1 transgenic mice as model systems, we firstly elucidated the mechanisms of interferon γ (IFNγ) regulation by PGE2 and PGI2. Specifically, PGE2 accumulation in astrocytes activated the ERK1/2 and NF-κB signaling pathways by phosphorylation, which resulted in IFNγ expression. In contrast, the administration of PGI2 attenuated the effects of PGE2 on stimulating the production of IFNγ via inhibiting the translocation of NF-κB from the cytosol to the nucleus. Due to these observations, we further studied these prostaglandins and found that both PGE2 and PGI2 increased Aβ1-42 levels. In detail, PGE2 induced IFNγ expression in an Aβ1-42-dependent manner, whereas PGI2-induced Aβ1-42 production did not alleviate cells from IFNγ inhibition by PGI2 treatment. More importantly, our data also revealed that not only Aβ1-42 oligomer but also fibrillar have the ability to induce the expression of IFNγ via stimulation of NF-κB nuclear translocation in astrocytes of APP/PS1 mice. The production of IFNγ finally accelerated the deposition of Aβ1-42 in β-amyloid plaques.
Figures
References
-
- Shoghi-Jadid K. et al. Localization of neurofibrillary tangles and beta-amyloid plaques in the brains of living patients with Alzheimer disease. Am J Geriatr Psychiatry 10, 24–35 (2002). - PubMed
-
- Heppner F. L., Ransohoff R. M. & Becher B. Immune attack: the role of inflammation in Alzheimer disease. Nat Rev Neurosci 16, 358–372 (2015). - PubMed
-
- Ford-Hutchinson A. W., Walker J. R., Davidson E. M. & Smith M. J. PGI2: a potential mediator of inflammation. Prostaglandins 16, 253–258 (1978). - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
