

BID Mediates Oxygen-Glucose Deprivation-Induced Neuronal Injury in Organotypic Hippocampal Slice Cultures and Modulates Tissue Inflammation in a Transient Focal Cerebral Ischemia Model without Changing Lesion Volume
- PMID: 26869884
- PMCID: PMC4737886
- DOI: 10.3389/fncel.2016.00014
BID Mediates Oxygen-Glucose Deprivation-Induced Neuronal Injury in Organotypic Hippocampal Slice Cultures and Modulates Tissue Inflammation in a Transient Focal Cerebral Ischemia Model without Changing Lesion Volume
Abstract
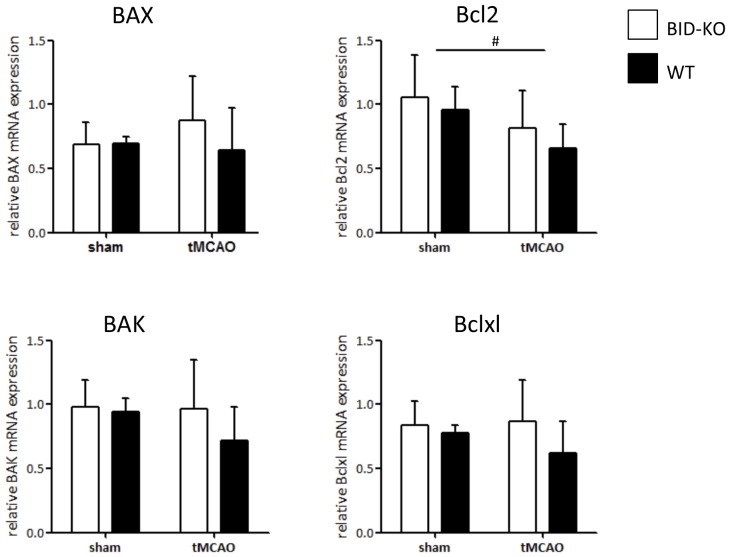
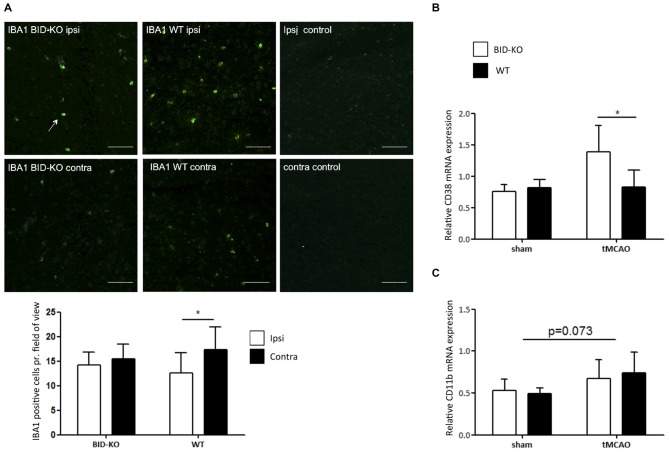
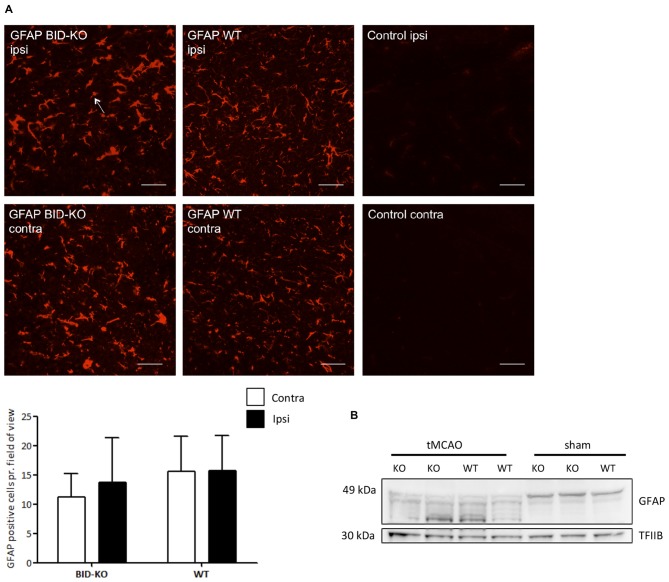
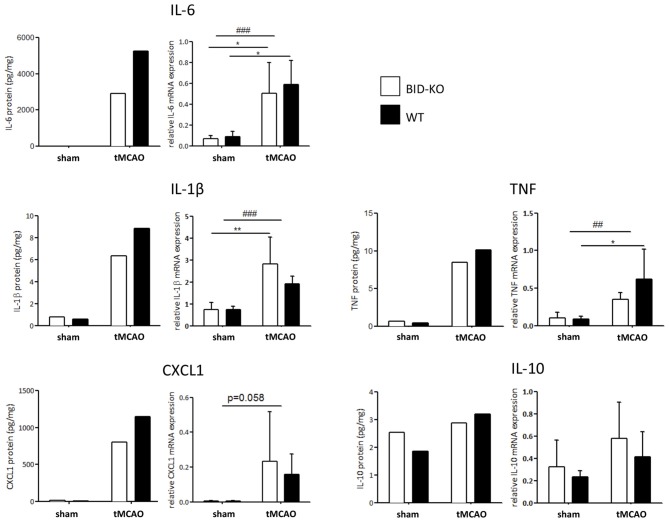
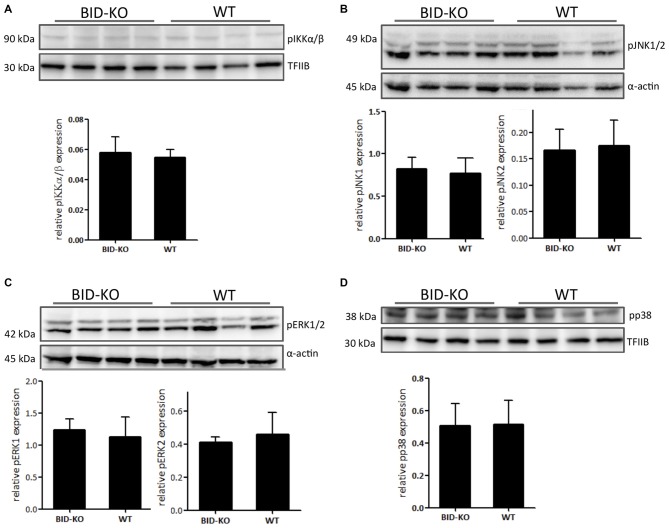
The BH3 interacting-domain death agonist (BID) is a pro-apoptotic protein involved in death receptor-induced and mitochondria-mediated apoptosis. Recently, it has also been suggested that BID is involved in the regulation of inflammatory responses in the central nervous system. We found that BID deficiency protected organotypic hippocampal slice cultures in vitro from neuronal injury induced by oxygen-glucose deprivation. In vivo, BID-knockout (KO) mice and wild type (WT) mice were subjected to 60 min of transient middle cerebral artery occlusion (tMCAO) to induce focal cerebral ischemia, and allowed to recover for 24 h. Infarct volumes and functional outcome were assessed and the inflammatory response was evaluated using immunofluorescence, Western blotting, quantitative PCR (qPCR) and Mesoscale multiplex analysis. We observed no difference in the infarct volume or neurological outcome between BID-KO and WT mice. The inflammatory response was reduced by BID deficiency as indicated by a change in microglial/leukocyte response. In conclusion, our data suggest that BID deficiency is neuroprotective in an in vitro model and modulates the inflammatory response to focal cerebral ischemia in vivo. However, this is not translated into a robust neuroprotection in vivo.
Keywords: BID; focal cerebral ischemia; inflammation; neuronal injury; organotypic hippocampal slice cultures.
Figures
Similar articles
-
BID mediates neuronal cell death after oxygen/ glucose deprivation and focal cerebral ischemia.Proc Natl Acad Sci U S A. 2001 Dec 18;98(26):15318-23. doi: 10.1073/pnas.261323298. Epub 2001 Dec 11. Proc Natl Acad Sci U S A. 2001. PMID: 11742085 Free PMC article.
-
Ionotropic glutamate receptors and glutamate transporters are involved in necrotic neuronal cell death induced by oxygen-glucose deprivation of hippocampal slice cultures.Neuroscience. 2005;136(3):779-94. doi: 10.1016/j.neuroscience.2005.07.020. Neuroscience. 2005. PMID: 16344151
-
Methods to induce primary and secondary traumatic damage in organotypic hippocampal slice cultures.Brain Res Brain Res Protoc. 2000 Apr;5(2):153-8. doi: 10.1016/s1385-299x(00)00007-6. Brain Res Brain Res Protoc. 2000. PMID: 10775835
-
Signaling of cell death and cell survival following focal cerebral ischemia: life and death struggle in the penumbra.J Neuropathol Exp Neurol. 2003 Apr;62(4):329-39. doi: 10.1093/jnen/62.4.329. J Neuropathol Exp Neurol. 2003. PMID: 12722825 Review.
-
Brain cooling during transient focal ischemia provides complete neuroprotection.Neurosci Biobehav Rev. 1997 Jan;21(1):31-44. doi: 10.1016/0149-7634(95)00080-1. Neurosci Biobehav Rev. 1997. PMID: 8994207 Review.
Cited by
-
PHACTR1 genetic variability is not critical in small vessel ischemic disease patients and PcomA recruitment in C57BL/6J mice.Sci Rep. 2021 Mar 16;11(1):6072. doi: 10.1038/s41598-021-84919-x. Sci Rep. 2021. PMID: 33727568 Free PMC article.
-
The loss-of-function disease-mutation G301R in the Na+/K+-ATPase α2 isoform decreases lesion volume and improves functional outcome after acute spinal cord injury in mice.BMC Neurosci. 2017 Sep 8;18(1):66. doi: 10.1186/s12868-017-0385-9. BMC Neurosci. 2017. PMID: 28886701 Free PMC article.
-
An exploratory investigation of brain collateral circulation plasticity after cerebral ischemia in two experimental C57BL/6 mouse models.J Cereb Blood Flow Metab. 2020 Feb;40(2):276-287. doi: 10.1177/0271678X19827251. Epub 2019 Feb 7. J Cereb Blood Flow Metab. 2020. PMID: 31549895 Free PMC article.
-
Neuronal Cell Death.Physiol Rev. 2018 Apr 1;98(2):813-880. doi: 10.1152/physrev.00011.2017. Physiol Rev. 2018. PMID: 29488822 Free PMC article. Review.
-
Neuronal Ablation of IKK2 Decreases Lesion Size and Improves Functional Outcome after Spinal Cord Injury in Mice.JSM Neurosurg Spine. 2017;5(3):1090. Epub 2017 Aug 10. JSM Neurosurg Spine. 2017. PMID: 30035210 Free PMC article.
References
-
- Bonner H. P., Concannon C. G., Bonner C., Woods I., Ward M. W., Prehn J. H. (2010). Differential expression patterns of Puma and Hsp70 following proteasomal stress in the hippocampus are key determinants of neuronal vulnerability. J. Neurochem. 114, 606–616. 10.1111/j.1471-4159.2010.06790.x - DOI - PubMed
-
- Carlson N. G., Wieggel W. A., Chen J., Bacchi A., Rogers S. W., Gahring L. C. (1999). Inflammatory cytokines IL-1 alpha, IL-1 beta, IL-6 and TNF-alpha impart neuroprotection to an excitotoxin through distinct pathways. J. Immunol. 163, 3963–3968. - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials