

Whole Genome Analysis of Injectional Anthrax Identifies Two Disease Clusters Spanning More Than 13 Years
- PMID: 26870786
- PMCID: PMC4740342
- DOI: 10.1016/j.ebiom.2015.10.004
Whole Genome Analysis of Injectional Anthrax Identifies Two Disease Clusters Spanning More Than 13 Years
Abstract
Background: Anthrax is a rare disease in humans but elicits great public fear because of its past use as an agent of bioterrorism. Injectional anthrax has been occurring sporadically for more than ten years in heroin consumers across multiple European countries and this outbreak has been difficult to trace back to a source.
Methods: We took a molecular epidemiological approach in understanding this disease outbreak, including whole genome sequencing of Bacillus anthracis isolates from the anthrax victims. We also screened two large strain repositories for closely related strains to provide context to the outbreak.
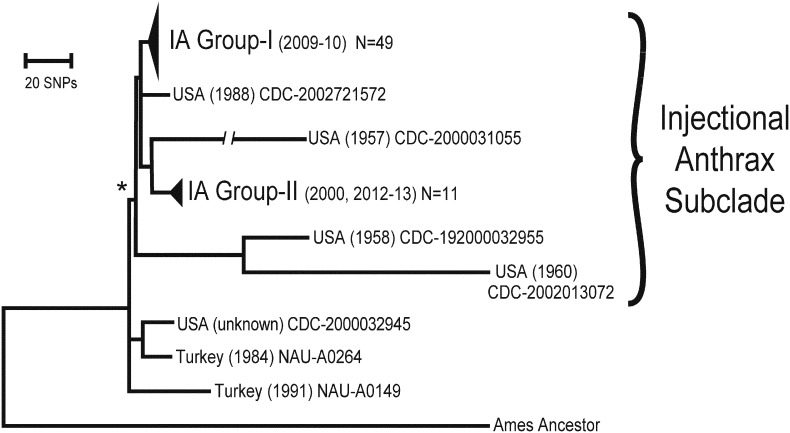
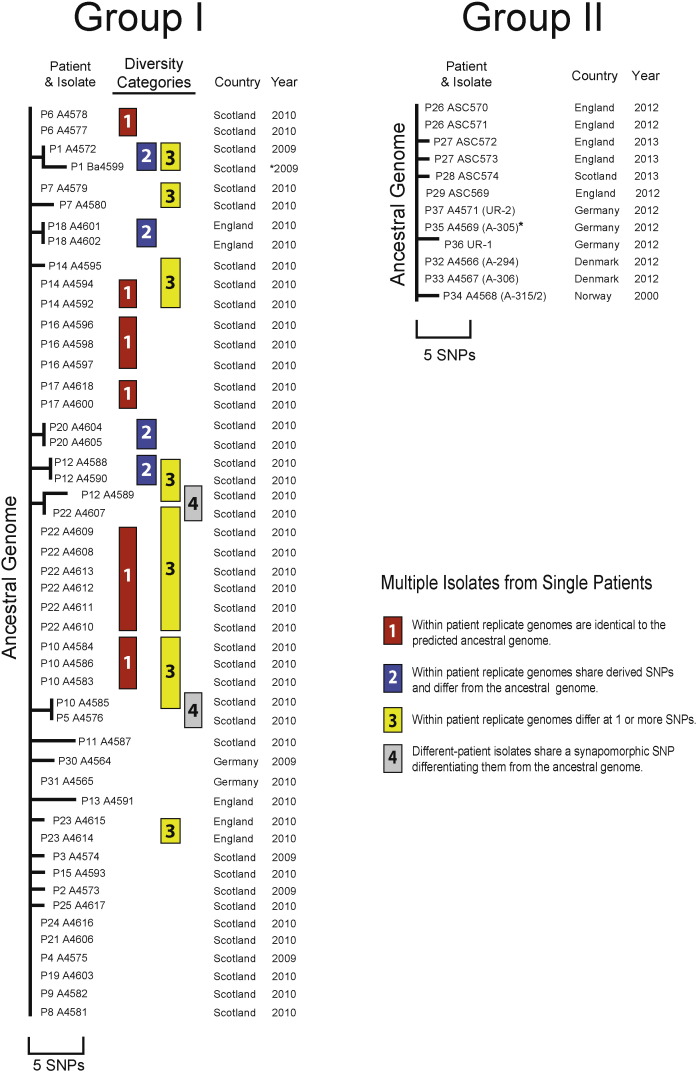
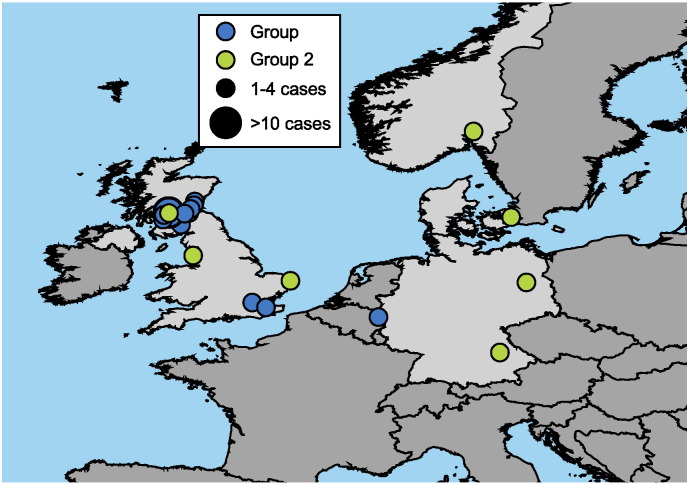
Findings: Analyzing 60 Bacillus anthracis isolates associated with injectional anthrax cases and closely related reference strains, we identified 1071 Single Nucleotide Polymorphisms (SNPs). The synapomorphic SNPs (350) were used to reconstruct phylogenetic relationships, infer likely epidemiological sources and explore the dynamics of evolving pathogen populations. Injectional anthrax genomes separated into two tight clusters: one group was exclusively associated with the 2009-10 outbreak and located primarily in Scotland, whereas the second comprised more recent (2012-13) cases but also a single Norwegian case from 2000.
Interpretation: Genome-based differentiation of injectional anthrax isolates argues for at least two separate disease events spanning > 12 years. The genomic similarity of the two clusters makes it likely that they are caused by separate contamination events originating from the same geographic region and perhaps the same site of drug manufacturing or processing. Pathogen diversity within single patients challenges assumptions concerning population dynamics of infecting B. anthracis and host defensive barriers for injectional anthrax.
Funding: This work was supported by the United States Department of Homeland Security grant no. HSHQDC-10-C-00,139 and via a binational cooperative agreement between the United States Government and the Government of Germany. This work was supported by funds from the German Ministry of Defense (Sonderforschungsprojekt 25Z1-S-431,214). Support for sequencing was also obtained from Illumina, Inc. These sources had no role in the data generation or interpretation, and had not role in the manuscript preparation.
Panel 1 research in context systematic review: We searched PubMed for any article published before Jun. 17, 2015, with the terms "Bacillus anthracis" and "heroin", or "injectional anthrax". Other than our previously published work (Price et al., 2012), we found no other relevant studies on elucidating the global phylogenetic relationships of B. anthracis strains associated with injectional anthrax caused by recreational heroin consumption of spore-contaminated drug. There were, however, publically available genome sequences of two strains involved (Price et al., 2012, Grunow et al., 2013) and the draft genome sequence of Bacillus anthracis UR-1, isolated from a German heroin user (Ruckert et al., 2012) with only limited information on the genotyping of closely related strains (Price et al., 2012, Grunow et al., 2013).
Lay person interpretation: Injectional anthrax has been plaguing heroin drug users across Europe for more than 10 years. In order to better understand this outbreak, we assessed genomic relationships of all available injectional anthrax strains from four countries spanning a > 12 year period. Very few differences were identified using genome-based analysis, but these differentiated the isolates into two distinct clusters. This strongly supports a hypothesis of at least two separate anthrax spore contamination events perhaps during the drug production processes. Identification of two events would not have been possible from standard epidemiological analysis. These comprehensive data will be invaluable for classifying future injectional anthrax isolates and for future geographic attribution.
Keywords: Bacillus anthracis; Heroin; Injectional Anthrax; Phylogeny; SNP; Whole Genome Sequencing.
Figures
Comment in
-
Improving Exploitation of Whole Genome Sequencing Data for Public Health, Forensic Microbiology and Biosafety.EBioMedicine. 2015 Nov 10;2(11):1566-7. doi: 10.1016/j.ebiom.2015.11.011. eCollection 2015 Nov. EBioMedicine. 2015. PMID: 26870766 Free PMC article. No abstract available.
References
-
- Berger T., Kassirer M., Aran A.A. Euro surveillance: Bulletin Europeen sur les maladies transmissibles = European Communicable Disease Bulletin. Vol. 19. 2014. Injectional anthrax — new presentation of an old disease. - PubMed
-
- Booth M.G., Hood J., Brooks T.J., Hart A. Health Protection Scotland Anthrax Clinical N. Anthrax infection in drug users. Lancet. 2010;375(9723):1345–1346. - PubMed
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
