

Collateral Lethality: A new therapeutic strategy in oncology
- PMID: 26870836
- PMCID: PMC4746004
- DOI: 10.1016/j.trecan.2015.10.002
Collateral Lethality: A new therapeutic strategy in oncology
Abstract
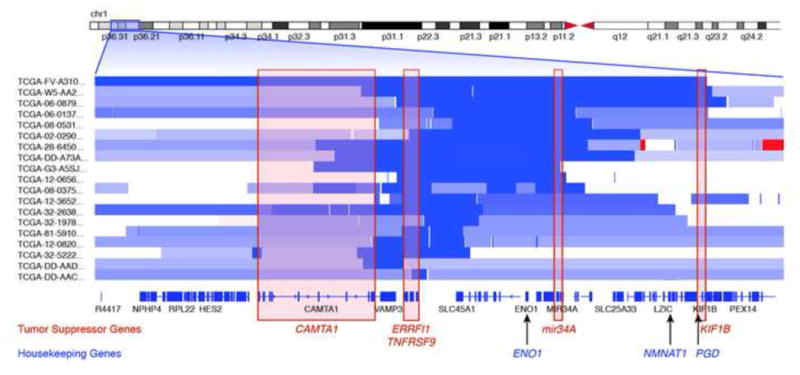
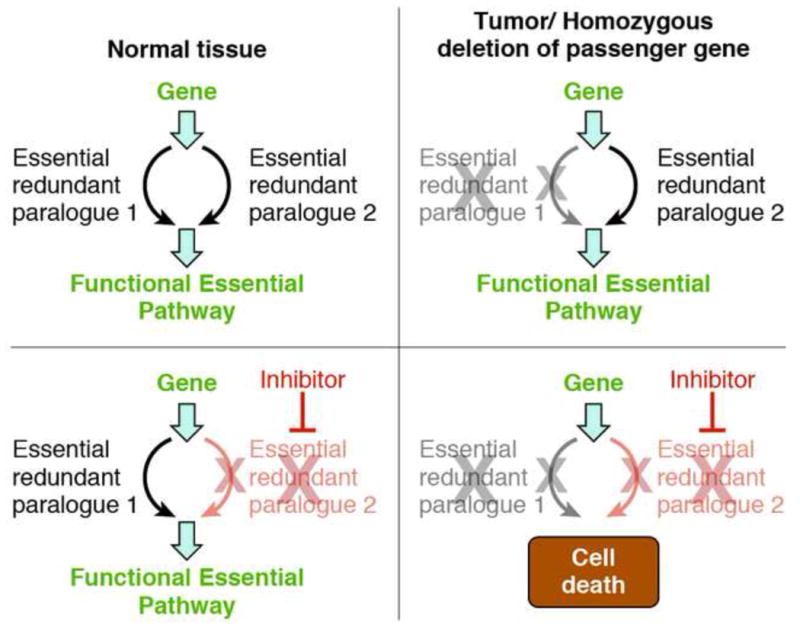
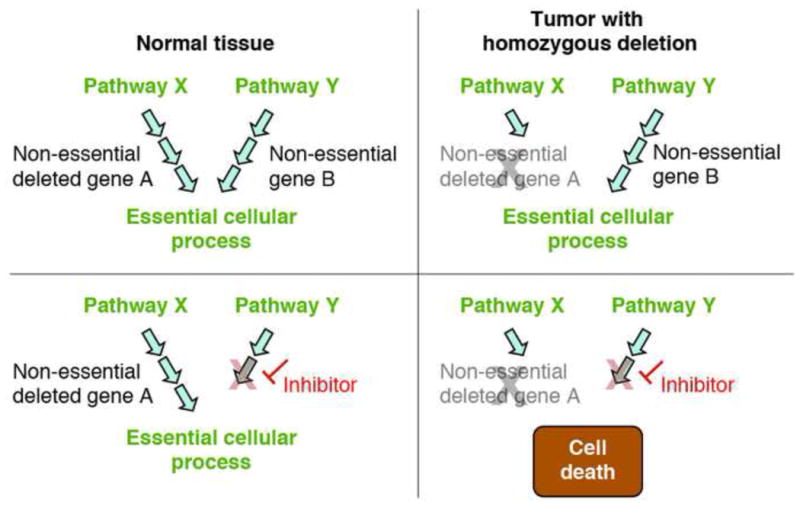
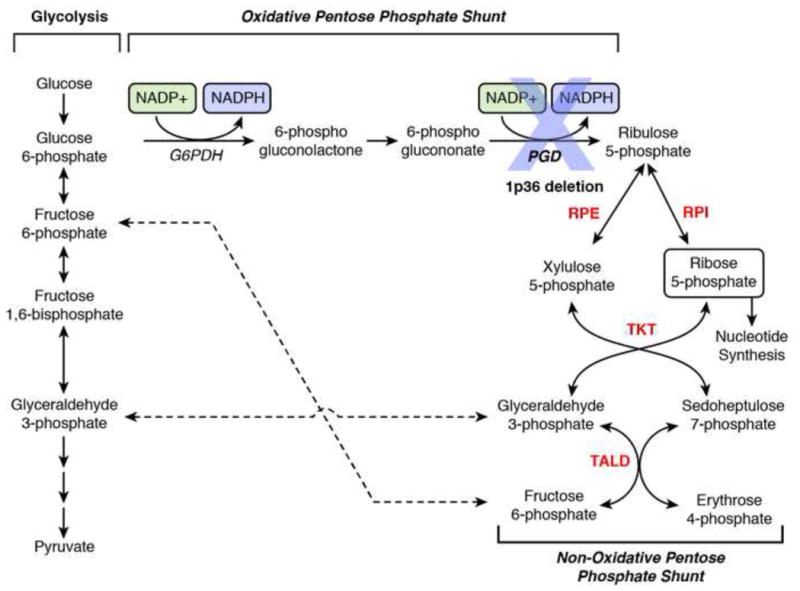
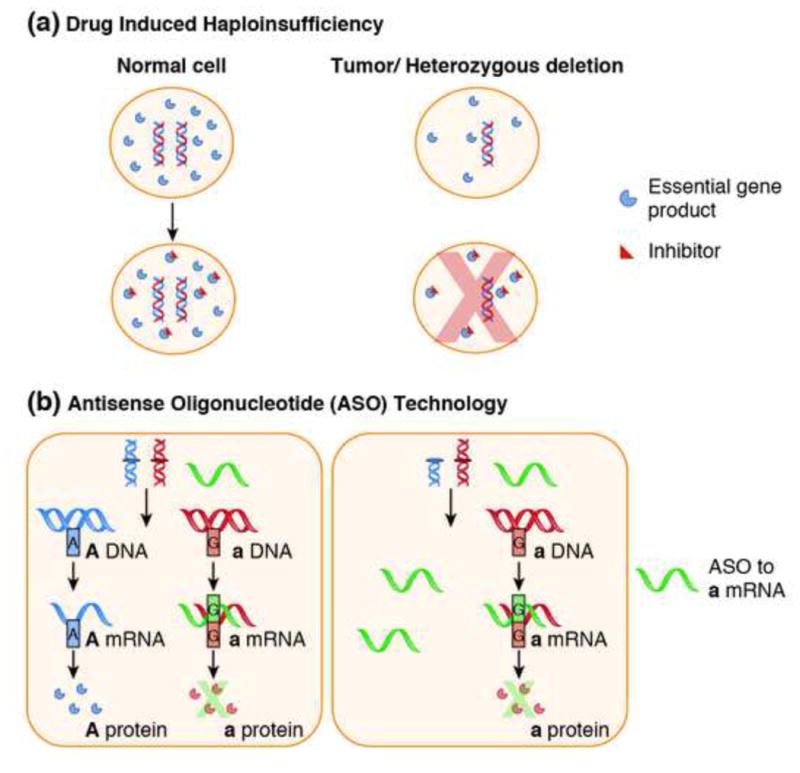
Genomic deletion of tumor suppressor genes (TSG) is a rite of passage for virtually all human cancers. The synthetic lethal paradigm has provided a framework for the development of molecular targeted therapeutics that are functionally linked to the loss of specific TSG functions. In the course of genomic events that delete TSGs, a large number of genes with no apparent direct role in tumor promotion also sustain deletion as a result of chromosomal proximity to the target TSG. In this perspective, we review the novel concept of "collateral lethality", which has served to identify cancer-specific therapeutic vulnerabilities resulting from co-deletion of passenger genes neighboring TSG. The large number of collaterally deleted genes, playing diverse functions in cell homeostasis, offers a rich repertoire of pharmacologically targetable vulnerabilities presenting novel opportunities for the development of personalized anti-neoplastic therapies.
Figures
References
-
- Arellano-Rodrigo E, Alvarez-Larran A. JAK inhibition in myelofibrosis. N Engl J Med. 2010;363(25):2464. author reply 2464-5; discussion 2465. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous