

Hepatitis C Virus Resistance to Carbohydrate-Binding Agents
- PMID: 26871442
- PMCID: PMC4752358
- DOI: 10.1371/journal.pone.0149064
Hepatitis C Virus Resistance to Carbohydrate-Binding Agents
Abstract
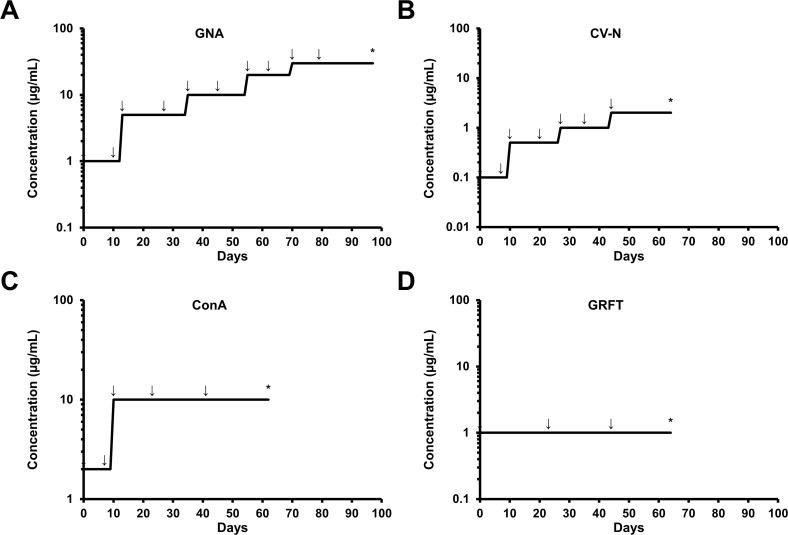
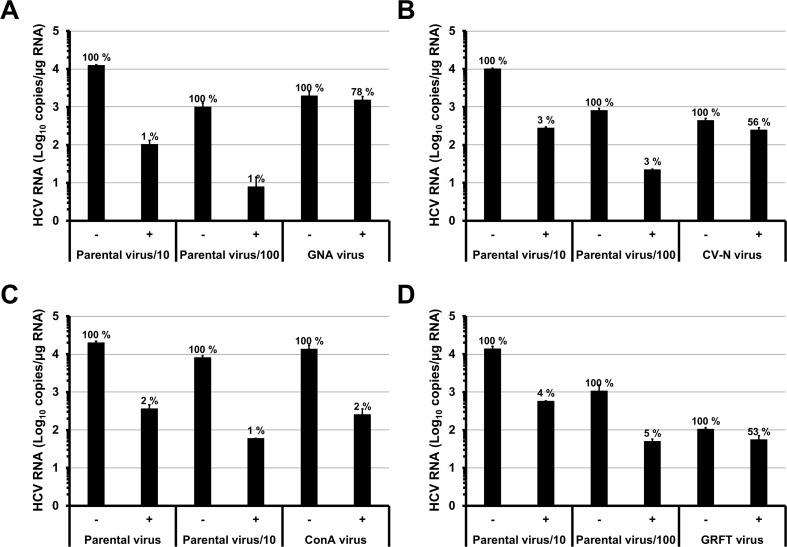
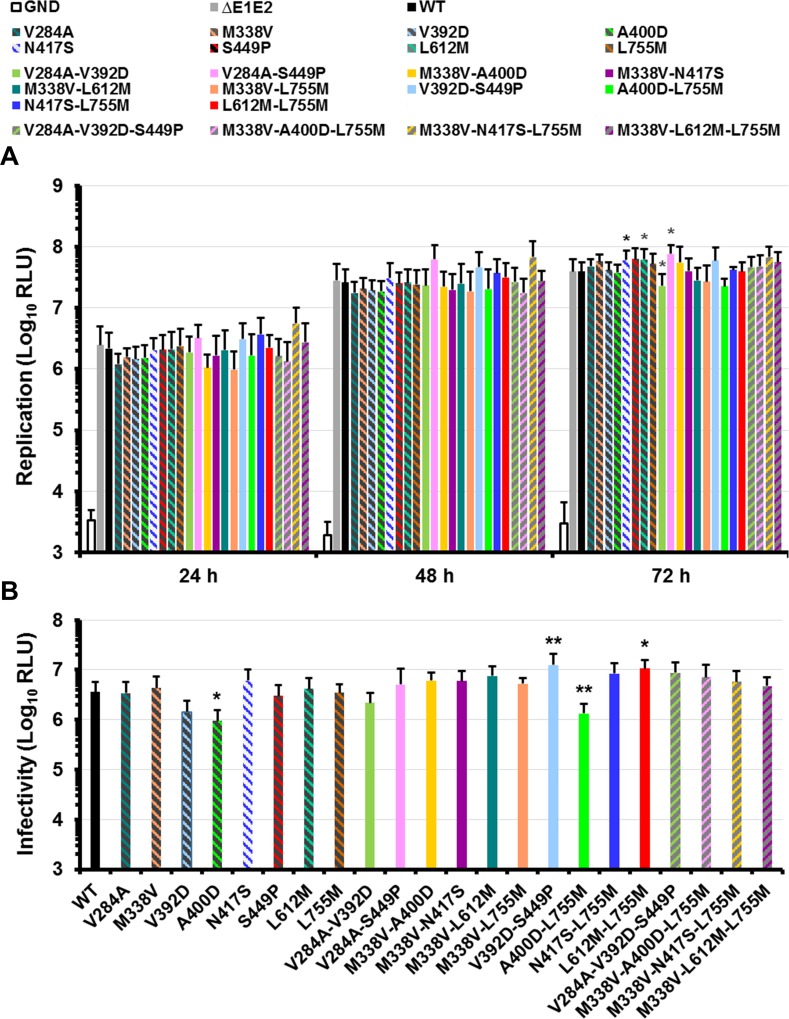
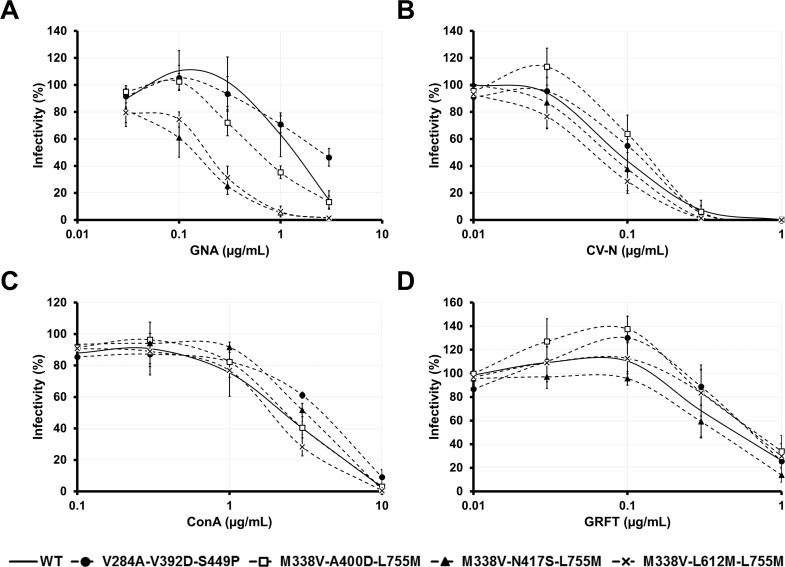
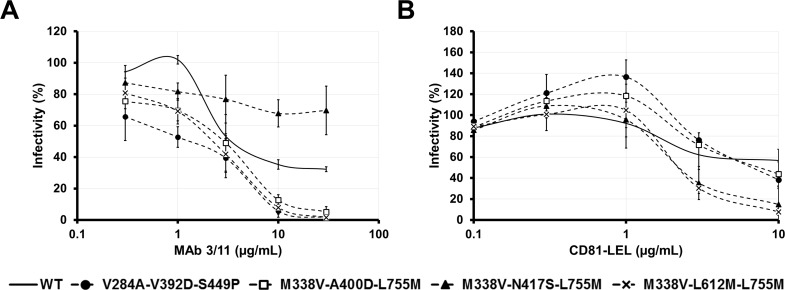
Carbohydrate binding agents (CBAs), including natural lectins, are more and more considered as broad-spectrum antivirals. These molecules are able to directly inhibit many viruses such as Human Immunodeficiency Virus (HIV), Hepatitis C Virus (HCV), Dengue Virus, Ebola Virus or Severe Acute Respiratory Syndrome Coronavirus through binding to envelope protein N-glycans. In the case of HIV, it has been shown that CBAs select for mutant viruses with N-glycosylation site deletions which are more sensitive to neutralizing antibodies. In this study we aimed at evaluating the HCV resistance to CBAs in vitro. HCV was cultivated in the presence of increasing Galanthus nivalis agglutinin (GNA), Cyanovirin-N, Concanavalin-A or Griffithsin concentrations, during more than eight weeks. At the end of lectin exposure, the genome of the isolated strains was sequenced and several potential resistance mutations in the E1E2 envelope glycoproteins were identified. The effect of these mutations on viral fitness as well as on sensitivity to inhibition by lectins, soluble CD81 or the 3/11 neutralizing antibody was assessed. Surprisingly, none of these mutations, alone or in combination, conferred resistance to CBAs. In contrast, we observed that some mutants were more sensitive to 3/11 or CD81-LEL inhibition. Additionally, several mutations were identified in the Core and the non-structural proteins. Thus, our results suggest that in contrast to HIV, HCV resistance to CBAs is not directly conferred by mutations in the envelope protein genes but could occur through an indirect mechanism involving mutations in other viral proteins. Further investigations are needed to completely elucidate the underlying mechanisms.
Conflict of interest statement
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
