

Frequent HRAS Mutations in Malignant Ectomesenchymoma: Overlapping Genetic Abnormalities With Embryonal Rhabdomyosarcoma
- PMID: 26872011
- PMCID: PMC4905780
- DOI: 10.1097/PAS.0000000000000612
Frequent HRAS Mutations in Malignant Ectomesenchymoma: Overlapping Genetic Abnormalities With Embryonal Rhabdomyosarcoma
Abstract
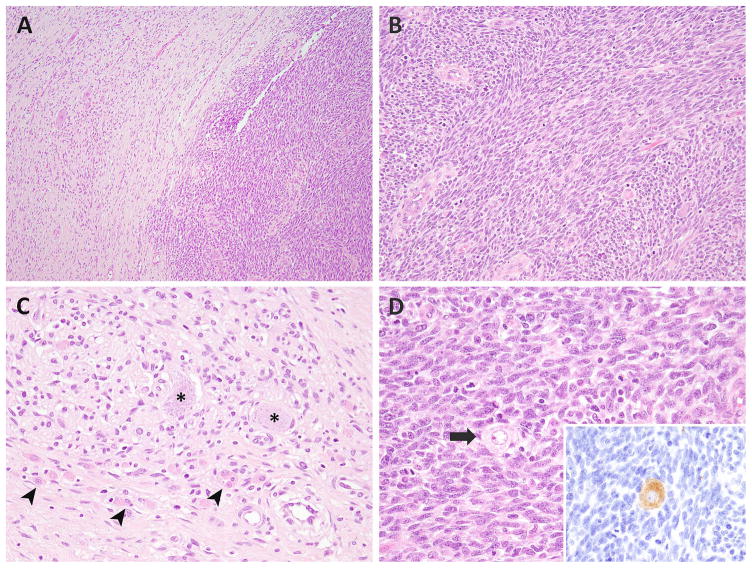
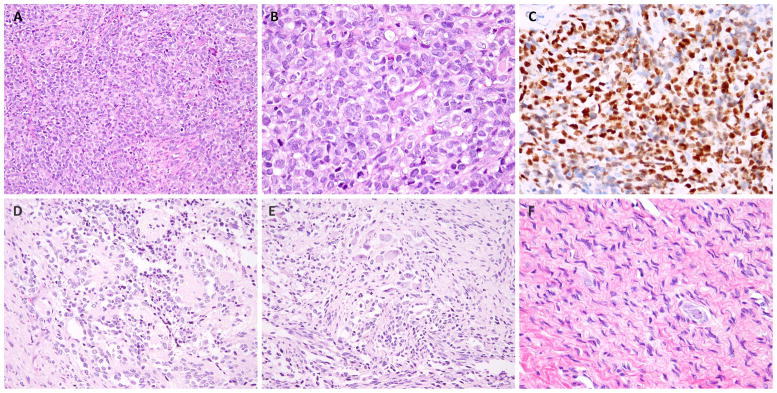
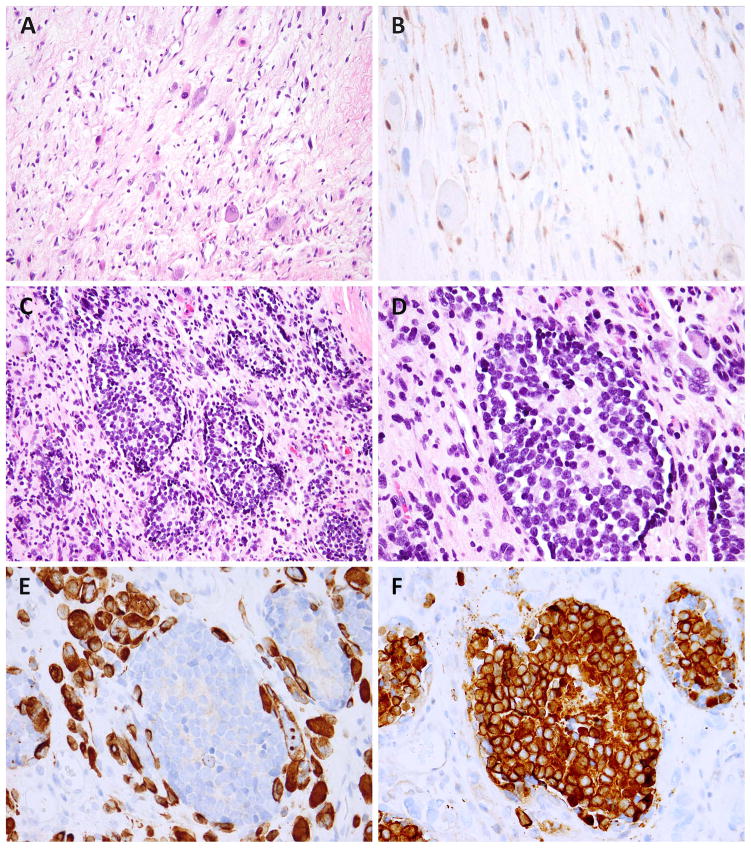
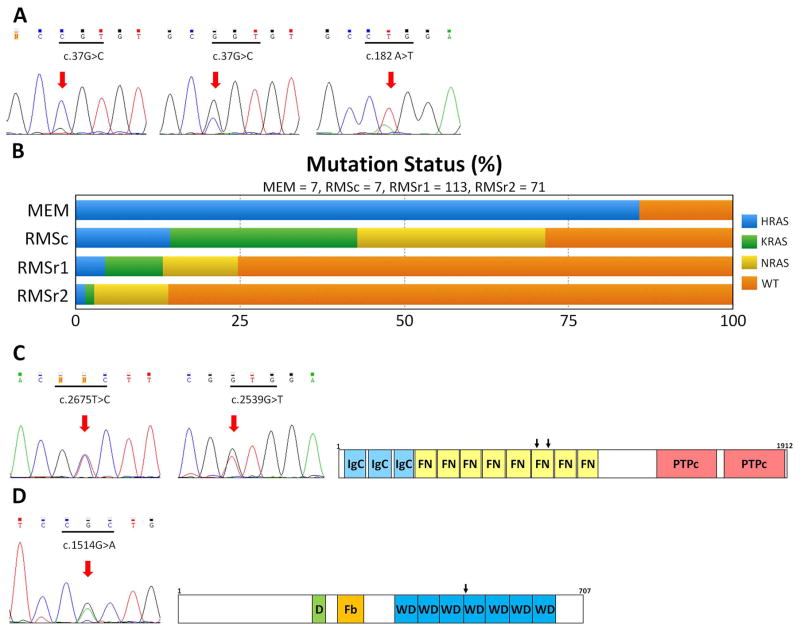
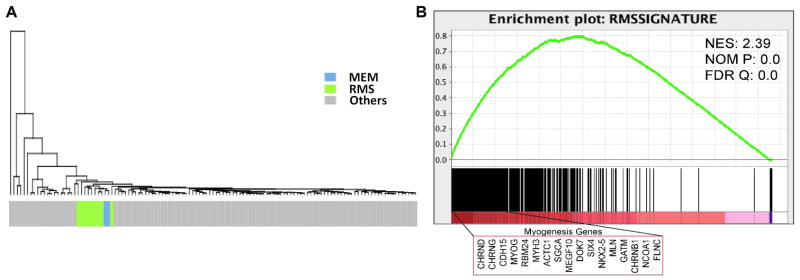
Malignant ectomesenchymoma (MEM) is an exceedingly rare pediatric sarcoma with a predilection for infants and young children and is composed of dual malignant mesenchymal and neuroectodermal components. Microscopically, MEM displays areas of rhabdomyosarcoma (RMS) with intermixed neuronal/neuroblastic foci. The molecular alterations associated with MEM and its relationship with embryonal RMS (ERMS) and malignant peripheral nerve sheath tumor (MPNST) have not yet been elucidated. In this study we used whole-transcriptome sequencing in 2 MEM index cases with available frozen tissue, followed by screening of the identified genetic abnormalities in 5 additional cases. No candidate fusion genes were detected by FusionSeq analysis; however, the mutation detection algorithms revealed HRAS and PTPRD hotspot mutations in both index cases, with 1 case harboring an additional FBXW7 mutation. As these mutation profiles have been previously described in ERMS we have tested their incidence in a control group of 7 age-matched ERMS. In addition, the gene signature of MEM was compared with that of RMS, MPNST, and neuronal lineage. All 7 MEM patients were male, with a mean age of 7.5 months (range, 0.6 to 17 mo). All except 1 occurred in the pelvic/urogenital region. Most cases showed ERMS elements, with occasional spindle or undifferentiated/round cell areas. The intermixed neuroectodermal components were mostly scattered ganglion cells, ganglioneuroma, or ganglioneuroblastoma. By Sanger sequencing, 6 of 7 (86%) MEMs had HRAS mutations, with no additional case harboring PTPRD or FBXW7 mutations. The only case lacking HRAS mutation showed neuroblastic micronodules without ganglion cells. The trimethylation at lysine 27 of histone H3 (H3K27me3) expression, typically lost in MPNST, was retained in all cases. In the control ERMS group, 5 of 7 (71%) showed RAS mutations, equally distributed among NRAS, KRAS, and HRAS genes. The expression profiling of MEM showed upregulation of skeletal muscle and neuronal genes, with no significant overlap with MPNST. Our results of common HRAS mutations and composite gene signature with RMS and neuronal/neuroblastic elements suggest a closer genetic link of MEM to RMS rather than to MPNST.
Figures
Similar articles
-
Molecular array analyses of 51 pediatric tumors shows overlap between malignant intracranial ectomesenchymoma and MPNST but not medulloblastoma or atypical teratoid rhabdoid tumor.Acta Neuropathol. 2007 Jun;113(6):695-703. doi: 10.1007/s00401-007-0210-0. Epub 2007 Mar 13. Acta Neuropathol. 2007. PMID: 17431644
-
Clinicopathologic and survival correlates of embryonal rhabdomyosarcoma driven by RAS/RAF mutations.Genes Chromosomes Cancer. 2022 Mar;61(3):131-137. doi: 10.1002/gcc.23010. Epub 2021 Nov 16. Genes Chromosomes Cancer. 2022. PMID: 34755412 Free PMC article.
-
Loss of H3K27me3 occurs in a large subset of embryonal rhabdomyosarcomas: Immunohistochemical and molecular analysis of 25 cases.Ann Diagn Pathol. 2021 Jun;52:151735. doi: 10.1016/j.anndiagpath.2021.151735. Epub 2021 Mar 22. Ann Diagn Pathol. 2021. PMID: 33770660
-
Rhabdomyosarcoma in children - current pathologic and molecular classification.Pol J Pathol. 2018;69(1):20-32. doi: 10.5114/pjp.2018.75333. Pol J Pathol. 2018. PMID: 29895123 Review.
-
An integrative morpho-molecular approach in malignant ectomesenchymoma diagnosis: report of a new paediatric case and a review of the literature.Front Oncol. 2024 Mar 1;14:1320541. doi: 10.3389/fonc.2024.1320541. eCollection 2024. Front Oncol. 2024. PMID: 38496756 Free PMC article. Review.
Cited by
-
First Reported Case of Malignant Ectomesenchymoma with p.Leu122Arg Mutation in MYOD1 Gene: Extensive Intra- and Extracranial Tumor in a 15-Year-Old Female.Head Neck Pathol. 2023 Sep;17(3):855-863. doi: 10.1007/s12105-023-01542-0. Epub 2023 Mar 13. Head Neck Pathol. 2023. PMID: 36913073 Free PMC article.
-
Somatic and mosaic HRAS mutations in pediatric malignant ectomesenchymoma.J Hum Genet. 2025 Jul 8. doi: 10.1038/s10038-025-01363-9. Online ahead of print. J Hum Genet. 2025. PMID: 40629179
-
Recurrent RET Gene Rearrangements in Intraductal Carcinomas of Salivary Gland.Am J Surg Pathol. 2018 Apr;42(4):442-452. doi: 10.1097/PAS.0000000000000952. Am J Surg Pathol. 2018. PMID: 29443014 Free PMC article.
-
Targeting RAS in pediatric cancer: is it becoming a reality?Curr Opin Pediatr. 2020 Feb;32(1):48-56. doi: 10.1097/MOP.0000000000000856. Curr Opin Pediatr. 2020. PMID: 31815779 Free PMC article. Review.
-
Congenital Malignant Ectomesenchymoma Presenting as a Neck Mass in a Newborn.Children (Basel). 2025 Apr 8;12(4):480. doi: 10.3390/children12040480. Children (Basel). 2025. PMID: 40310147 Free PMC article.
References
-
- Naka A, Matsumoto S, Shirai T, et al. Ganglioneuroblastoma associated with malignant mesenchymoma. Cancer. 1975;36:1050–1056. - PubMed
-
- Karcioglu Z, Someren A, Mathes SJ. Ectomesenchymoma. A malignant tumor of migratory neural crest (ectomesenchyme) remnants showing ganglionic, schwannian, melanocytic and rhabdomyoblastic differentiation. Cancer. 1977;39:2486–2496. - PubMed
-
- Freitas AB, Aguiar PH, Miura FK, et al. Malignant ectomesenchymoma. Case report and review of the literature. Pediatr Neurosurg. 1999;30:320–330. - PubMed
-
- Kawamoto EH, Weidner N, Agostini RM, Jr, et al. Malignant ectomesenchymoma of soft tissue. Report of two cases and review of the literature. Cancer. 1987;59:1791–1802. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
