

Compensatory Increase of Transglutaminase 2 Is Responsible for Resistance to mTOR Inhibitor Treatment
- PMID: 26872016
- PMCID: PMC4752276
- DOI: 10.1371/journal.pone.0149388
Compensatory Increase of Transglutaminase 2 Is Responsible for Resistance to mTOR Inhibitor Treatment
Expression of concern in
-
Expression of concern: Compensatory increase of transglutaminase 2 is responsible for resistance to mTOR inhibitor treatment.PLoS One. 2020 Jan 15;15(1):e0227851. doi: 10.1371/journal.pone.0227851. eCollection 2020. PLoS One. 2020. PMID: 31940416 Free PMC article. No abstract available.
Abstract
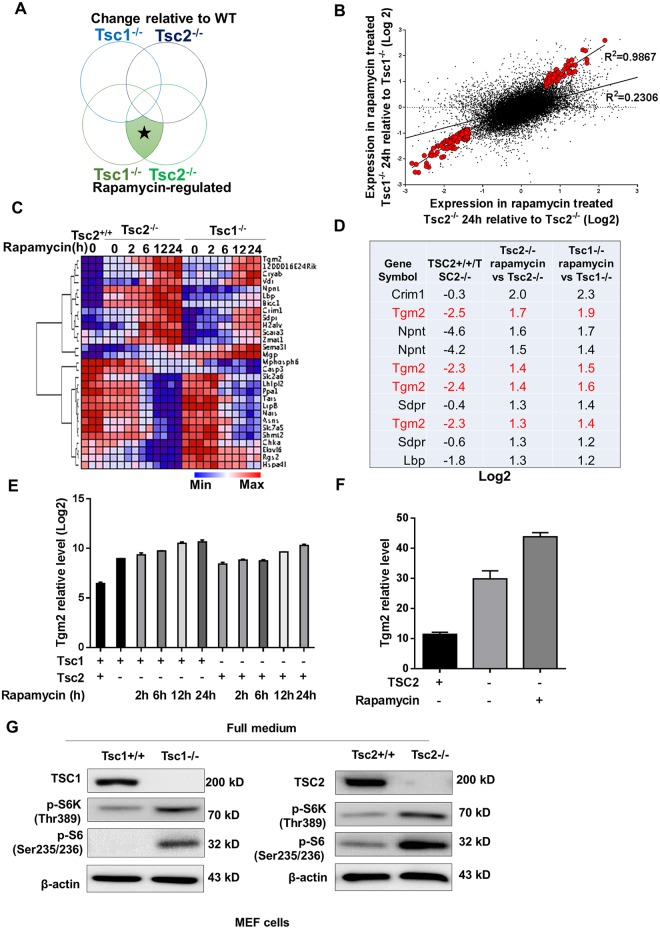
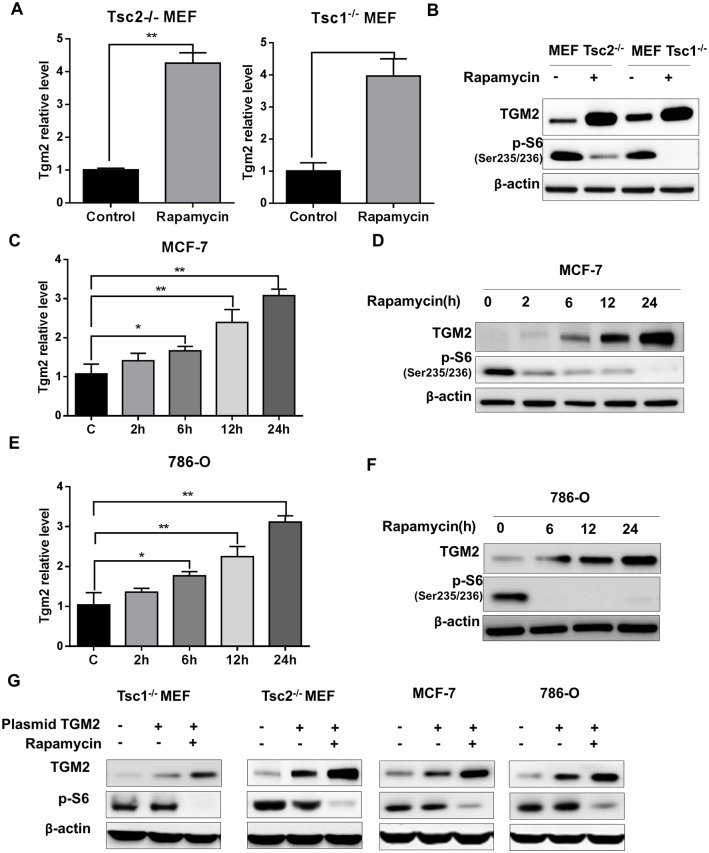
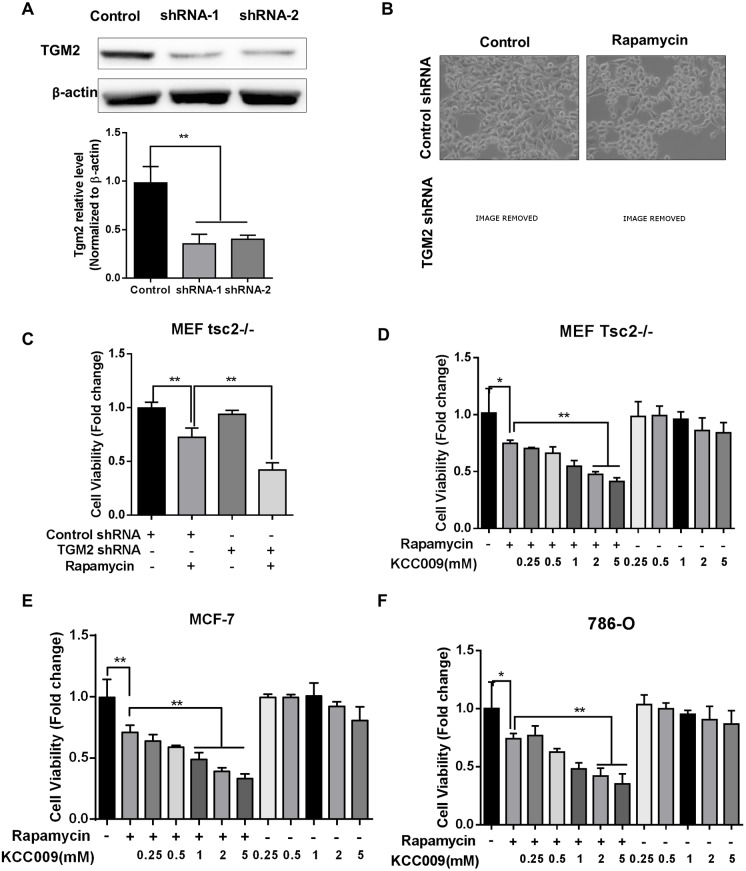
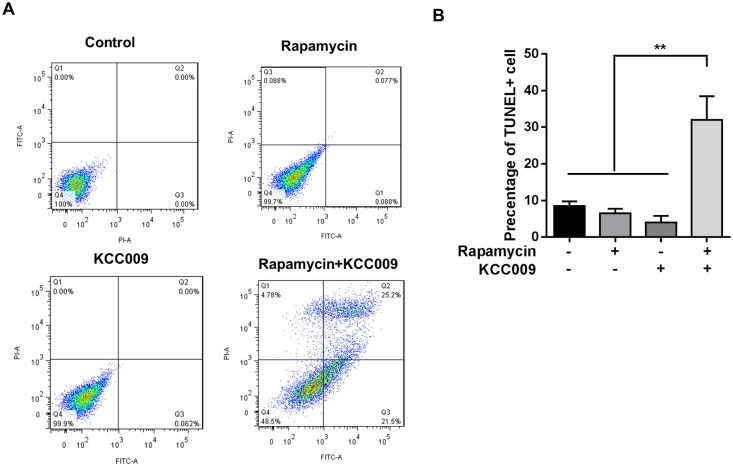
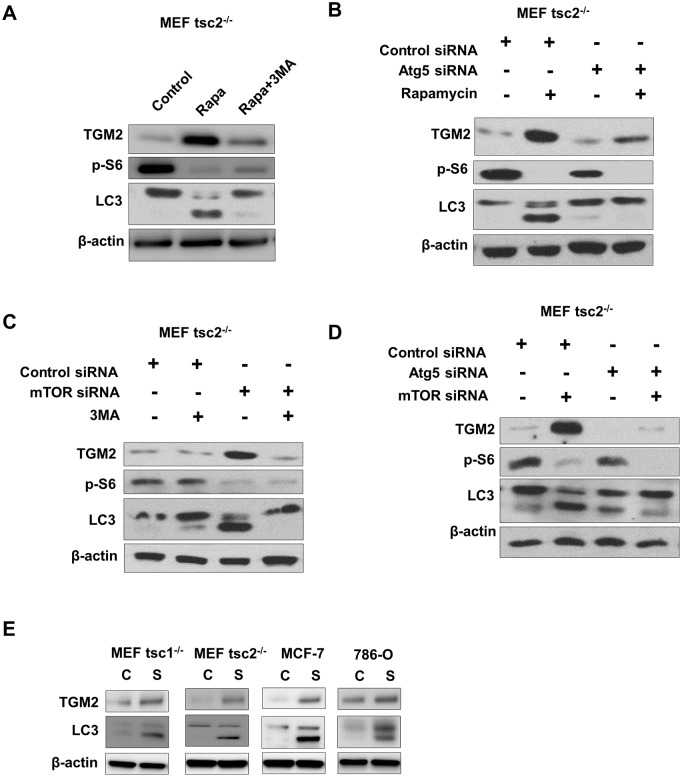
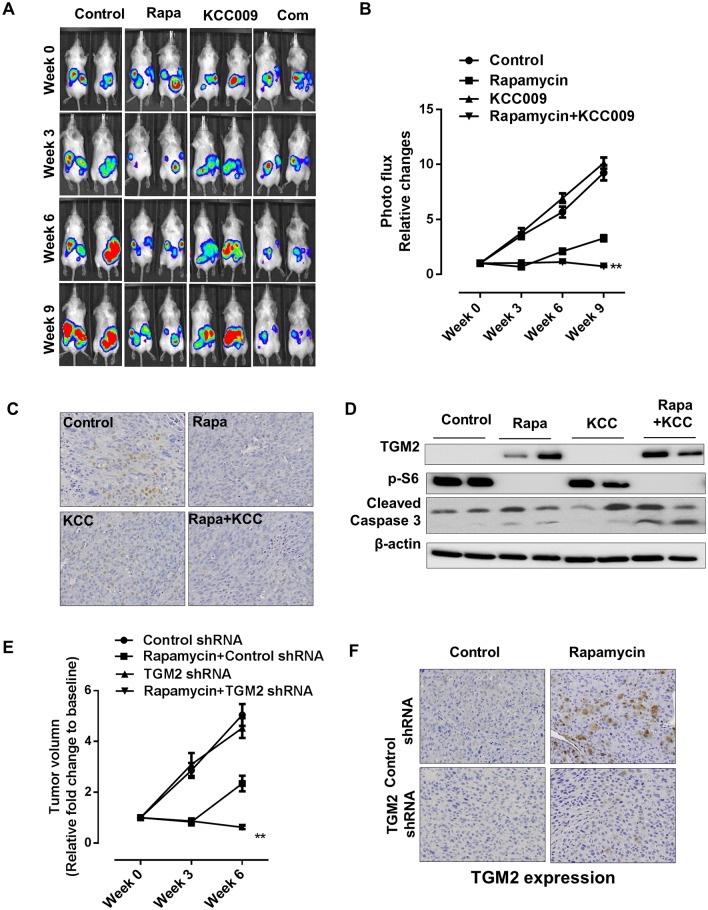
The mechanistic target of rapamycin complex 1 (mTORC1) plays a crucial role in controlling cell growth and homeostasis. Deregulation of mTOR signaling is frequently observed in some cancers, making it an attractive drug target for cancer therapy. Although mTORC1 inhibitor rapalog-based therapy has shown positive results in various pre-clinical animal cancer studies, tumors rebound upon treatment discontinuation. Moreover, several recent clinical trials showed that the mTORC1 inhibitors rapamycin and rapalog only reduce the capacity for cell proliferation without promoting cell death, consistent with the concept that rapamycin is cytostatic and reduces disease progression but is not cytotoxic. It is imperative that rapamycin-regulated events and additional targets for more effective drug combinations be identified. Here, we report that rapamycin treatment promotes a compensatory increase in transglutaminase 2 (TGM2) levels in mTORC1-driven tumors. TGM2 inhibition potently sensitizes mTORC1-hyperactive cancer cells to rapamycin treatment, and a rapamycin-induced autophagy blockade inhibits the compensatory TGM2 upregulation. More importantly, tumor regression was observed in MCF-7-xenograft tumor-bearing mice treated with both mTORC1 and TGM2 inhibitors compared with those treated with either a single inhibitor or the vehicle control. These results demonstrate a critical role for the compensatory increase in transglutaminase 2 levels in promoting mTORC1 inhibitor resistance and suggest that rational combination therapy may potentially suppress cancer therapy resistance.
Conflict of interest statement
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
