

Pseudouridines in U2 snRNA stimulate the ATPase activity of Prp5 during spliceosome assembly
- PMID: 26873591
- PMCID: PMC4801943
- DOI: 10.15252/embj.201593113
Pseudouridines in U2 snRNA stimulate the ATPase activity of Prp5 during spliceosome assembly
Abstract
Pseudouridine (Ψ) is the most abundant internal modification identified in RNA, and yet little is understood of its effects on downstream reactions. Yeast U2 snRNA contains three conserved Ψs (Ψ35, Ψ42, and Ψ44) in the branch site recognition region (BSRR), which base pairs with the pre-mRNA branch site during splicing. Here, we show that blocks to pseudouridylation at these positions reduce the efficiency of pre-mRNA splicing, leading to growth-deficient phenotypes. Restoration of pseudouridylation at these positions using designer snoRNAs results in near complete rescue of splicing and cell growth. These Ψs interact genetically with Prp5, an RNA-dependent ATPase involved in monitoring the U2 BSRR-branch site base-pairing interaction. Biochemical analysis indicates that Prp5 has reduced affinity for U2 snRNA that lacks Ψ42 and Ψ44 and that Prp5 ATPase activity is reduced when stimulated by U2 lacking Ψ42 or Ψ44 relative to wild type, resulting in inefficient spliceosome assembly. Furthermore, in vivo DMS probing analysis reveals that pseudouridylated U2, compared to U2 lacking Ψ42 and Ψ44, adopts a slightly different structure in the branch site recognition region. Taken together, our results indicate that the Ψs in U2 snRNA contribute to pre-mRNA splicing by directly altering the binding/ATPase activity of Prp5.
Keywords: Prp5 ATPase; U2 snRNA; pseudouridylation; spliceosome assembly; splicing.
© 2016 The Authors.
Figures

Schematic representation of U2‐pre‐
mRNA interaction. Partial sequences/secondary structures of vertebrate and Saccharomyces cerevisiae U2 are schematically shown. Also shown are the base‐pairing interactions between the U2 branch site recognition region (BSRR ) and the pre‐mRNA branch site (BS ). Pseudouridines (Ψ) are indicated. Enzymes responsible for yeast U2 pseudouridylation at the three conserved sites are also indicated.Temperature‐sensitive growth assay. Yeast strains (wild‐type or deletion mutants, indicated on the left) were transformed with either an empty vector or a plasmid carrying an artificial guide
RNA gene targeting U2 pseudouridylation at positions 42 and 44 (indicated on the top), and grown at various temperatures (indicated at the bottom).Blockage of pseudouridylation by deletion of specific pseudouridylase genes.
RNA s isolated from wild‐type and pseudouridylase‐deletion strains (indicated on the top) were used for U2 pseudouridylation assay (CMC modification followed by primer extension). Primer extension pauses/stops correspond to Ψ sites (indicated on the left and right). Note: The above‐background signal of Ψ42 in the snr81 lane (and to some extent, in the snr81 pus7 lane) is likely caused by the presence of strong Ψ44 band. This is a primer extension artifact that often occurs when there is a strong Ψ signal in the neighboring position. Alternatively, it is possible that having Ψ at position 44 allows for an unidentified enzyme other than snR81 to modify U42 (albeit inefficiently; potentially Pus1, which is responsible for Ψ44 formation, can act in a “processive” manner). Lanes 1–8 and lanes 9–16 are from two separate gels.Restoration of U2 pseudouridylation by an artificial box H/
ACA RNA .RNA s isolated from various yeast strains (indicated on the top), including the snr81Δ pusΔ strain that was transformed with an artificial guideRNA targeting positions 42 and 44 (lanes 9 and 10), were assayed for pseudouridylation (see legend to C).
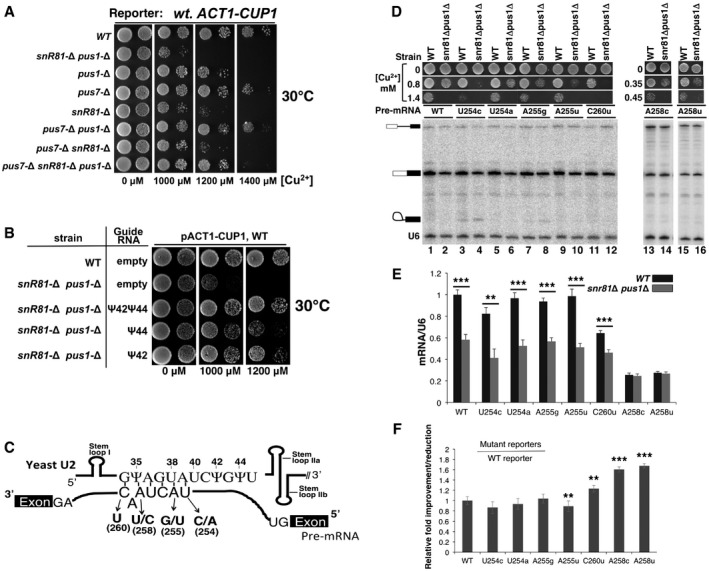
Growth assay using the
ACT 1‐CUP 1 reporter system. Yeast cells carrying theACT 1‐CUP 1 reporter were deleted of pseudouridylase genes (indicated on the left) and were then assayed for growth at 30°C on media containing various concentrations of [Cu2+] (indicated at the bottom).Growth rescue by restoration of pseudouridylation. The wild‐type strain (from A) or the snr81Δ pusΔ strain (also from A), which exhibited the most severe growth‐deficiency phenotype, was transformed with an empty vector or a plasmid carrying a pseudouridylation guide
RNA gene targeting either position 42, position 44, or both (indicated). The resulting cells were assayed for growth at 30°C on media containing various concentrations of [Cu2+] (indicated at the bottom).Schematic representation of U2–pre‐
mRNA interaction. The U2BSRR sequence and the pre‐mRNA branch site sequence are shown. The arrows indicate several point mutations at the pre‐mRNA branch site. Stem loops I,II a, andII b are also indicated.Splicing assay using the wild‐type and mutant
ACT 1‐CUP 1 reporters.RNA s isolated from the wild‐type and the snr81Δ pusΔ strains (indicated on the top), which carry the wild‐typeACT 1‐CUP 1 reporter pre‐mRNA or any of the mutantACT 1‐CUP 1 reporter pre‐mRNA s (indicated, see also C), were used for splicing assay (primer extension analysis). The un‐spliced pre‐mRNA , lariat intermediate, and splicedmRNA are indicated. A primer complementary to U6 was also used (as an internal control) in the assay, and the U6 band is indicated as well. In addition, the growth phenotype of each strain (in various concentrations of [Cu2+]) is also shown. Lanes 1–12, lanes 13 and 14, and lanes 15 and 16 are from separate gels.Quantification of
ACT 1‐CUP 1mRNA levels. SplicedACT 1‐CUP 1mRNA levels were calculated relative to U6 in each lane. The quantification was based on three independent experiments.Relative improvement/reduction for the mutant reporters. Relative improvement or reduction in splicing was calculated by normalizing the splicing efficiency (
mRNA /U6) of each mutant reporter to that ofWT reporter (set at 1).

Prp5‐U2 co‐immunoprecipitation (
IP ). Wild‐type (lanes 1 and 2) and snR81Δ pus1Δ (lane 3) strains, each containing aFLAG ‐taggedPRP 5 gene (indicated on the top), were used for anti‐FLAG IP in the absence (lane 1) or presence (lanes 2 and 3) ofATP . The precipitated Prp5 (WT Prp5‐FLAG ) and co‐precipitated U1 and U2, detected by primer extension, are indicated.Quantification of co‐precipitated U2 sn
RNA . For each lane (theWT lane and the snr81Δ pus1Δ lane, corresponding to lane 2 and lane 3 of A, respectively), Prp5‐co‐precipitated U2 was normalized against co‐precipitated U1 [(intensity of the U2 band)/(intensity of the U1 band)]. The snr81Δ pus1Δ lane was then further normalized against theWT lane [(the value of normalized U2 from the snr81Δ pus1Δ lane)/(the value of normalized U2 from theWT lane)]. Quantification was based on three independent experiments.U2 expression level in wild‐type and snR81Δ pus1Δ strains. Total
RNA , isolated from the wild‐type (lane 1) or snR81Δ pus1Δ (lane 2) strains, was used for primer extension to measure the levels of U2 and U6. The U2 and U6 bands are indicated.Depletion of Prp5. Wild‐type (lanes 1, 3, and 4) and snR81Δ pus1Δ (lanes 2 and 5) cells containing a
FLAG ‐taggedPRP 5 were lysed, and anti‐FLAG IP was performed at a high concentration of salt. Cell extracts, before (lanes 1 and 2) and after [lanes 3 (mock), 4 and 5] anti‐FLAG IP , were used for Western analysis using anti‐FLAG . The Prp5 band is indicated.IP of reconstituted Prp5 (GAR )‐U2. An equal amount ofFLAG ‐tagged Prp5 (GAR ) was added to the wild‐type (lane 1) and snR81Δ pus1Δ (lane 2) cell extracts depleted of endogenous Prp5 (see C). After the salt concentration was brought back to its original level, anti‐FLAG IP was carried out. The Prp5 (GAR ) band, detected by Western, is indicated. The co‐precipitated U1 and U2 bands, detected by primer extension, are also indicated.Quantification of U2 that is co‐precipitated with mutant Prp5 (
GAR ). For each lane (including theWT lane and the snr81Δ pus1Δ lane, corresponding to lane 1 and lane 2 of E, respectively), U2 co‐precipitated with Prp5 (GAR ) was normalized against co‐precipitated U1 [(intensity of the U2 band)/(intensity of the U1 band)]. The snr81Δ pus1Δ lane was then further normalized against theWT lane [(the value of normalized U2 from the snr81Δ pus1Δ lane)/(the value of normalized U2 from theWT lane)]. Quantification was based on three independent experiments.Prp5‐U2‐pre‐
mRNA co‐IP . Reconstitution was carried out as described in (D). Prior toIP , an equal amount of radiolabeled pre‐mRNA was also added to each reaction. Anti‐FLAG IP was then performed in the presence ofATP . The precipitated Prp5 (WT andGAR ) band, detected by Western, is indicated. The co‐precipitated pre‐mRNA , directly visualized after electrophoresis, is also indicated. The pre‐mRNA input is also shown. In lanes 1 and 2, wild‐type and snR81Δ pus1Δ cell extracts, depleted of Prp5, were reconstituted with the wild‐typeFLAG ‐Prp5. In lanes 3 and 4, wild‐type and snR81Δ pus1Δ cell extracts, depleted of Prp5, were reconstituted with mutantFLAG ‐Prp5 (GAR ).Quantification of pre‐
mRNA co‐precipitated with Prp5 and U2. For each lane in (G), pre‐mRNA co‐precipitated with Prp5 (wild‐type orGAR ) was normalized against the Prp5 (WT /GAR ) band in the same lane (detected by anti‐FLAG ) [(intensity of pre‐mRNA )/(intensity ofWT /GAR Prp5)]. Lanes 2, 3, and 4 (see G) were then further normalized against lane 1 (see G) [(the value of normalized pre‐mRNA from lane 2, 3, or 4 in G)/(the value of normalized pre‐mRNA from lane 1 in G)]. The numbers at the bottom of the bar graph correspond to the lane numbers in (G). Quantification was based on three independent experiments.

Prp5‐U2 co‐immunoprecipitation (
IP ). Wild‐type (lane 1) and snR81Δ pus1Δ (lane 2) strains, each containing aFLAG ‐taggedWT PRP 5 gene (indicated on the top), were lysed, and the extracts from the two strains were each divided into two equal aliquots. One aliquot was directly used for anti‐FLAG IP at a low concentration of salt (50 mMKC l) (lanes 1 and 2). The other aliquot was used for anti‐FLAG IP at a high concentration of salt (1 MKC l), thus depleting the extracts ofWT Prp5 (lanes 3 and 4). Subsequently,FLAG ‐tagged mutant Prp5 (GAR ) were added back to the extracts, and salt concentration was brought back to the original concentration by dialysis. Anti‐FLAG IP was subsequently performed (lane 3, derived fromWT ; and lane 4, from snR81Δ pus1Δ). The co‐precipitated U1 and U2 in each sample were detected by primer extension using an equal amount of the same primers (anti‐U1 and anti‐U2 primers).Quantification of co‐precipitated U2 sn
RNA . For each lane [lane 1,WT U2 andWT Prp5; lane 2, U2 lacking Ψ42 and Ψ44 andWT Prp5; lane 3,WT U2 and mutant Prp5 (GAR ); lane 4, U2 lacking Ψ42 and Ψ44 and mutant Prp5 (GAR )], Prp5‐co‐precipitated U2 was normalized against co‐precipitated U1 [(intensity of the U2 band)/(intensity of the U1 band)]. All lanes were then further normalized against lane 1 (WT U2 andWT Prp5) [(the value of normalized U2 from each lane)/(the value of normalized U2 from lane 1)]. Quantification was based on three independent experiments. Error bars, mean ± SD. **P < 0.01, ***P < 0.001 (Student's t‐test).

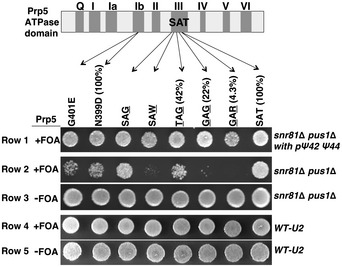
Prp5's
ATP ase activity assay using synthetic U2. The 5' sequence of Saccharomyces cerevisiae U2 is shown (top). The three Ψs (35, 42, and 44) and the branch site recognition sequence (underlined) are indicated. In theATP ase assay (bottom), noRNA (lane 3), or an equal amount of LiCl‐precipitatedRNA (lane 1) ortRNA (lane 2), a short U2 fragment (nts 1–76) (lane 4), a fully pseudouridylated short U2 fragment (nts 1–76) (lane 5), a long U2 fragment (nts 1–120) (lane 6), or a fully pseudouridylated long U2 fragment (nts 1–120) (lane 7), was used. The un‐hydrolyzedATP and hydrolyzed productADP are indicated. Lanes 1 and 2, lanes 3–5, and lanes 6 and 7 are from separate gels.Quantification of Prp5's
ATP ase activity activated by in vitro transcribed U2. Prp5'sATP ase activity shown in (A) was calculated using the formula (ADP )/(ADT +ATP ). The numbers at the bottom of the bar graph correspond to the lane numbers in (A). Quantification was based on three independent experiments.U2‐Prp5 binding assay.
FLAG ‐Prp5 and a radiolabeled U2 fragment (nts 1–76), either un‐pseudouridylated (lanes 1 and 2) or pseudouridylated (lanes 3 and 4), were incubated under the conditions used forATP ase activity assay. Anti‐FLAG IP was then carried out. Co‐precipitated (lanes 2 and 4) and un‐precipitated (lanes 1 and 3) U2 RNAs were analyzed by electrophoresis. The U2 band is indicated.Quantification of Prp5‐U2 binding. Relative intensities of U2 bands shown in (C) were calculated [setting the un‐precipitated uridine‐containing U2 (lane 1 and 5) to 1]. The numbers at the bottom of the bar graph correspond to the lane numbers in (C). Quantification was based on three independent experiments.
Prp5's
ATP ase activity assay using cellular U2. U2RNA isolated from the wild‐type strain (lanes 2 and 4) or from the snR81Δ pus1Δ strain (lanes 6 and 8) was used for theATP ase activity assay. In odd‐numbered lanes, noRNA was added. Both the wild‐type Prp5 (lanes 1–4) and the mutant Prp5 (GAR ) (lanes 5–8) were tested.Quantification of Prp5's
ATP ase activity activated by U2 from cells. Prp5'sATP ase activity shown in (E) was calculated using the formula (ADP )/(ADT +ATP ). The numbers at the bottom of the bar graph correspond to the lane numbers in (E). Quantification was based on four independent experiments.U2 level in the
ATP ase activity reactions.RNA s from lanes 2 and 4 in (C) were recovered, and U2 level was measured using primer extension. Lanes 1‐5, a titration of U2 snRNA . Lane 6, U2 from the wild‐type strain (lane 2 in C). Lane 7, U2 from the snR81Δ pus1Δ strain (lane 4 in C).
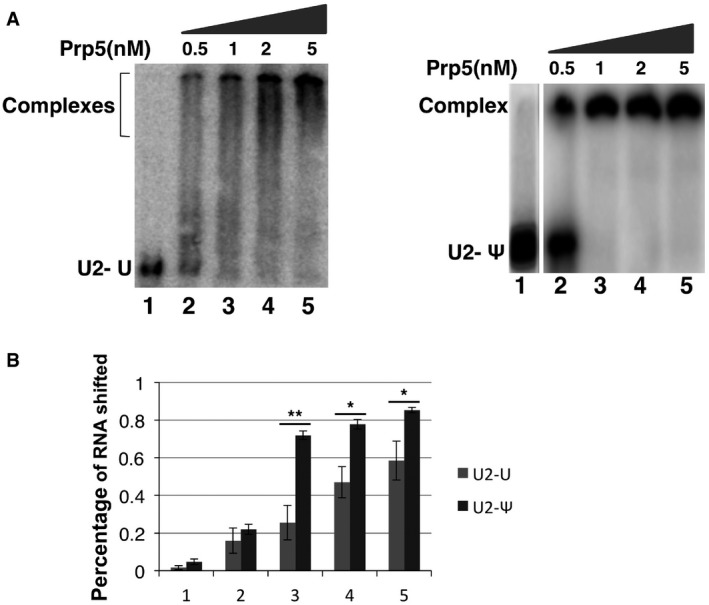
Prp5‐U2 gel‐shift assay. In vitro transcription was used to synthesize a radiolabeled and un‐pseudouridylated 5' fragment of U2 and a radiolabeled and pseudouridylated 5' fragment of U2 (
UTP was replaced by ΨTP during transcription). A small fixed amount of U2 fragment was used for Prp5‐U2 gel‐shift assay. The un‐pseudouridylated U2 fragment (left panel) and the pseudouridylated U2 fragment were each mixed with 0 (lane 1), 0.5, 1, 2, and 5 nM ofWT Prp5 (right panel). After a ∼10‐min incubation in a low‐salt buffer, the reactions were loaded onto a 5% native polyacrylamide gel, electrophoresed at a low voltage, and autoradiographed. Lane 1 and lanes 2‐5 are from two separate gels. Interestingly, while Prp5‐pseudouridylated U2 shifted nicely and efficiently (right panel), the Prp5‐un‐pseudouridylated U2 complex bands smeared (left panel). It is possible that un‐pseudouridylated U2 adopted alternative folding structures (with similar G), allowing Prp5 to bind (perhaps simultaneously but loosely) at multiple sites (non‐specific binding sites). On the other hand, incorporation of pseudouridines may rigidify or stabilize the most favorable U2 structure (Charette & Gray, 2000; Ge & Yu, 2013), promoting efficient and site‐specific binding with Prp5. It is worth noting that the conditions used forIP [binding at low‐salt concentration (50 mMKC l), and washing with a buffer of higher concentration of salt (150 mM), see Materials and Methods] were more stringent than the conditions used for gel‐shift assay (binding at 50 mMKC l, and no washing). Thus, the loosely bound complexes between Prp5 and un‐pseudouridylated U2 might be detected by gel‐shift assay but not byIP (see Fig 5C and D). Nonetheless, the gel‐shift results indicate that Prp5 has a higher binding affinity for pseudouridylated U2 than for un‐pseudouridylated U2 (quantified in B). The results from both assays (co‐IP and gel‐shift) are highly consistent.Quantification of gel‐shifted Prp5‐U2 complexes. The percentage of Prp5‐U2 complexes detected in (A) was calculated [(intensity of the complex band)/(intensity of the complex band + intensity of U2 band)]. Quantification was based on three independent experiments. Error bars, mean ± SD. *P < 0.05, **P < 0.01 (Student's t‐test).
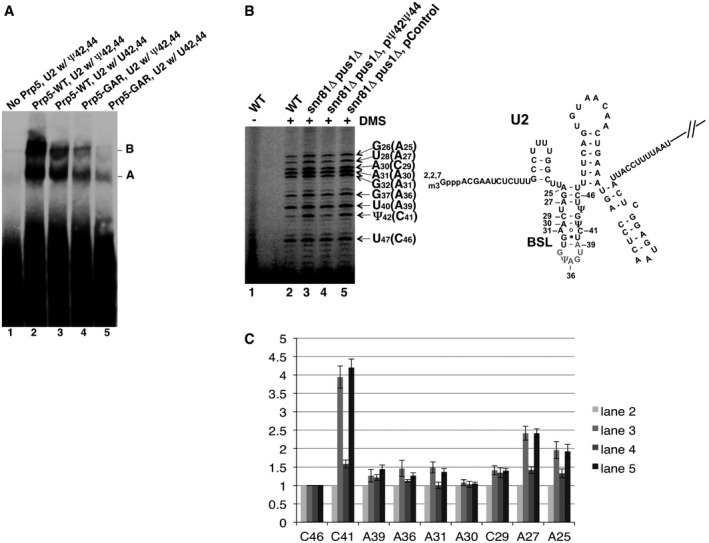
Native gel analysis of pre‐splicing complexes. Pre‐splicing complex assembly was carried out in the test tube with Prp5‐reconstituted cell extracts and labeled pre‐
mRNA . Lane 1, Prp5‐depleted wild‐type cell extract. Lane 2, Prp5‐depleted wild‐type cell extract, plus wild‐type Prp5. Lane 3, Prp5‐depleted snR81Δ pus1Δ cell extract, plus wild‐type Prp5. Lane 4, Prp5‐depleted wild‐type cell extract, plus mutant Prp5 (GAR ). Lane 5, Prp5‐depleted snR81Δ pus1Δ cell extract, plus mutant Prp5 (GAR ). The pre‐splicing complexes A and B are indicated.DMS in vivo probing. After being exposed toDMS , yeast cells were lysed, totalRNA collected, and primer extension analysis carried out (left panel). Lane 1, cells that were not exposed toDMS . Lane 2, wild‐type cells exposed toDMS . Lane 3, snR81Δ pus1Δ cells exposed toDMS . Lane 4, snR81Δ pus1Δ cells, transformed with a plasmid carrying an artificial guideRNA gene targeting positions 42 and 44, and exposed toDMS . Lane 5, snR81Δ pus1Δ cells, transformed with a plasmid carrying a control guideRNA gene with random guide sequences, and exposed toDMS . The nucleotides in parentheses areDMS ‐modified C and A residues that are one nucleotide after the actual primer extension stops (indicated by the arrows). A partial U2 sequence (along with theBSL structure) is shown (right panel).DMS ‐modified adenosine and cytosine in theBSL are also indicated.Residue accessibility quantification. Relative accessibility (relative band intensity) was quantified based on three independent experiments. The intensity of every band in each lane was normalized against the C46 band (the least changed band; compare lanes 2–5 in B) in its respective lane [(intensity of a band)/(intensity of C46 in the same lane)]. Each C46‐normalized band in lanes 3–5 (refer to B) was then further normalized against its respective counterpart in lane 2 [(the value of a C46‐normalized band in lanes 3–5)/(the value of its counterpart in lane 2)]. The error bars represent the standard deviations of the measurements (based on three independent experiments).

References
-
- Arnez JG, Steitz TA (1994) Crystal structure of unmodified tRNA(Gln) complexed with glutaminyl‐tRNA synthetase and ATP suggests a possible role for pseudo‐uridines in stabilization of RNA structure. Biochemistry 33: 7560–7567 - PubMed
-
- Bakin AV, Ofengand J (1998) Mapping of pseudouridine residues in RNA to nucleotide resolution. Methods Mol Biol 77: 297–309 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
