

Actin dynamics and cofilin-actin rods in alzheimer disease
- PMID: 26873625
- PMCID: PMC5345344
- DOI: 10.1002/cm.21282
Actin dynamics and cofilin-actin rods in alzheimer disease
Abstract
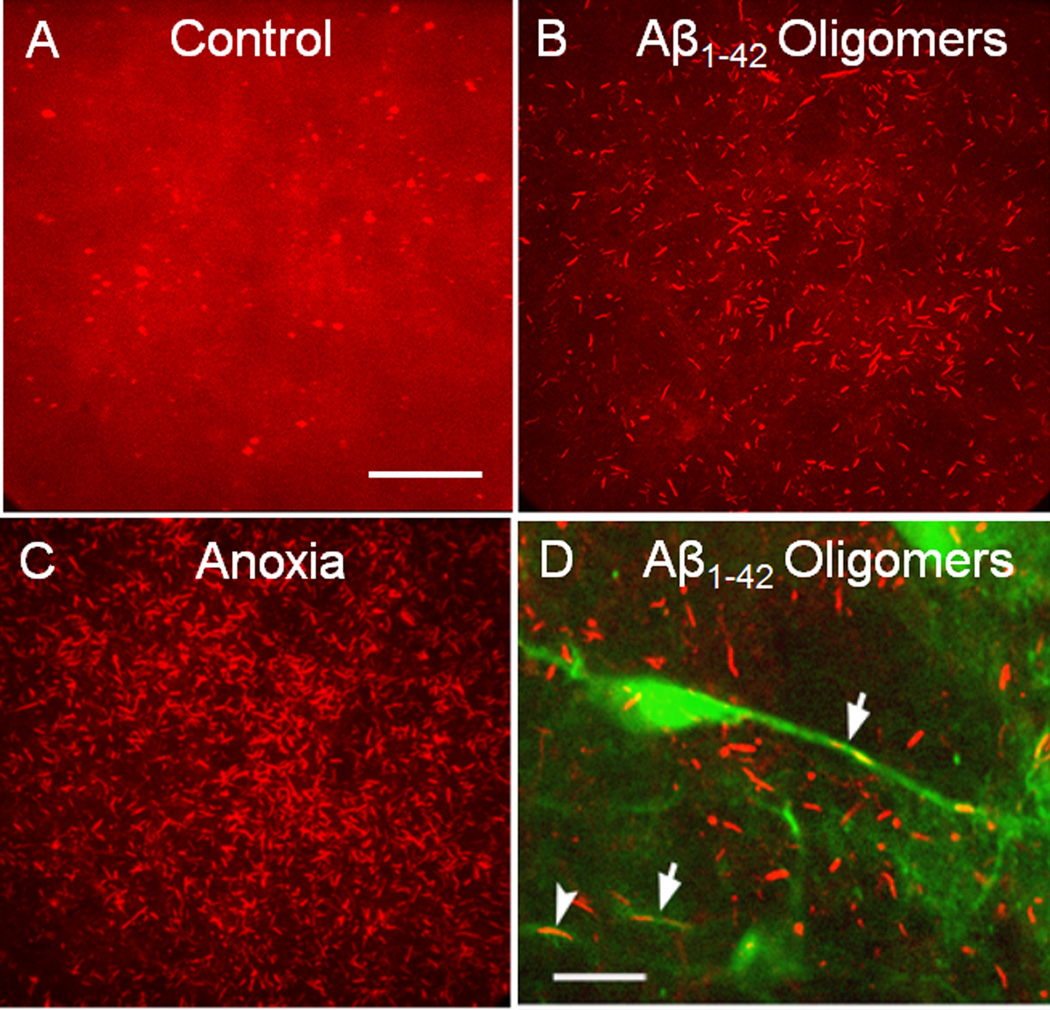
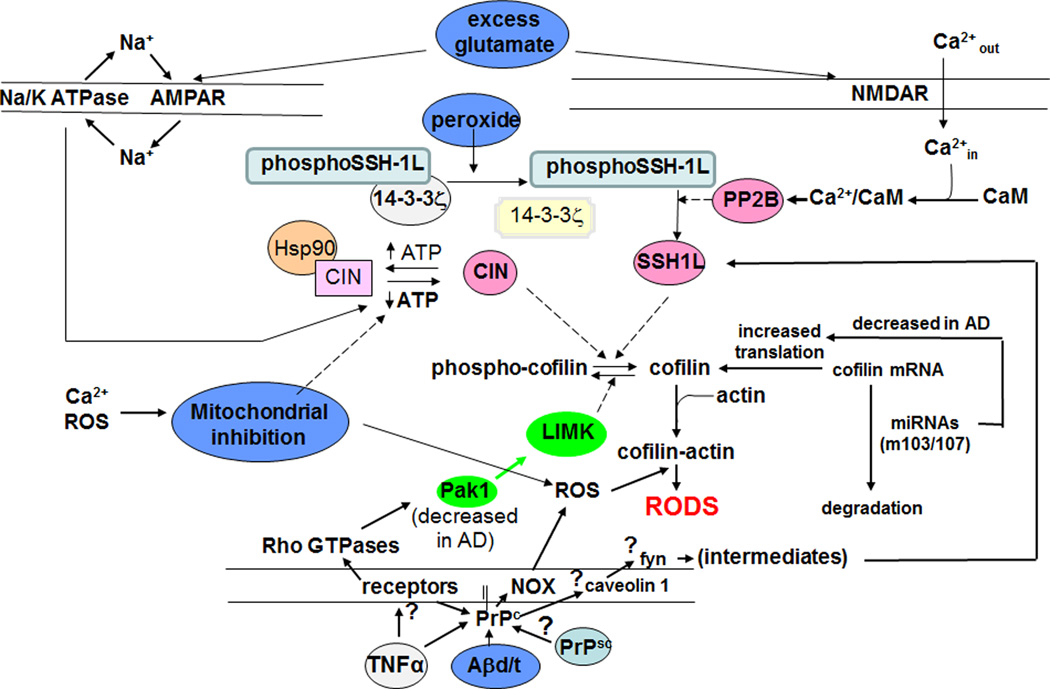
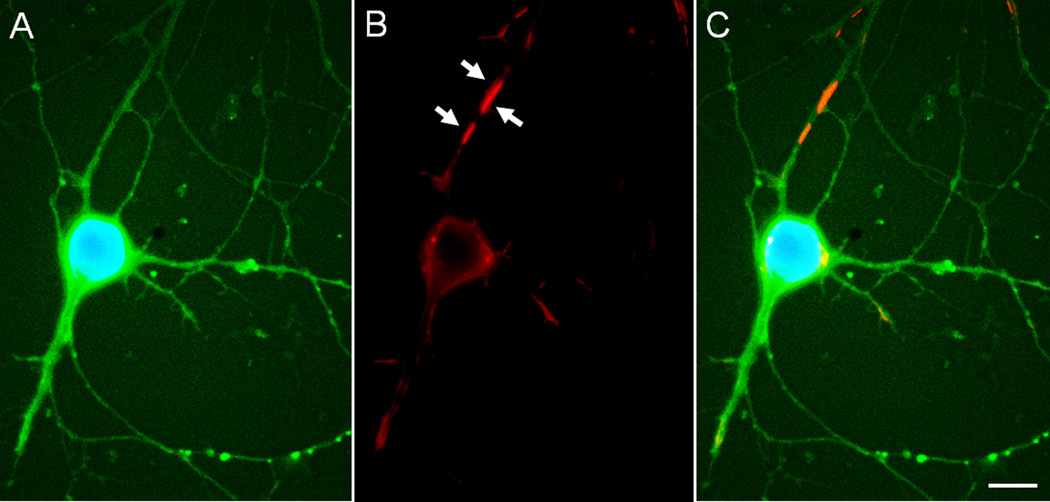
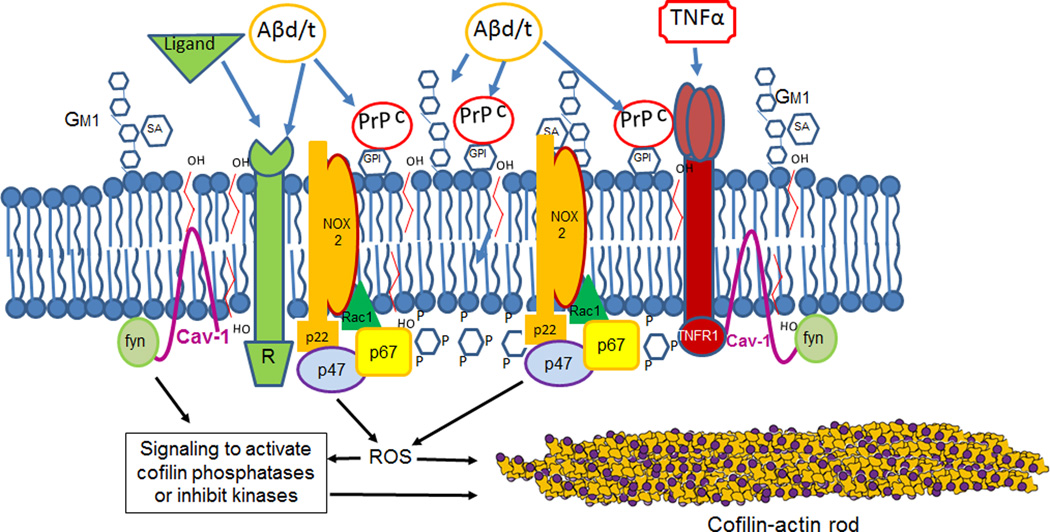
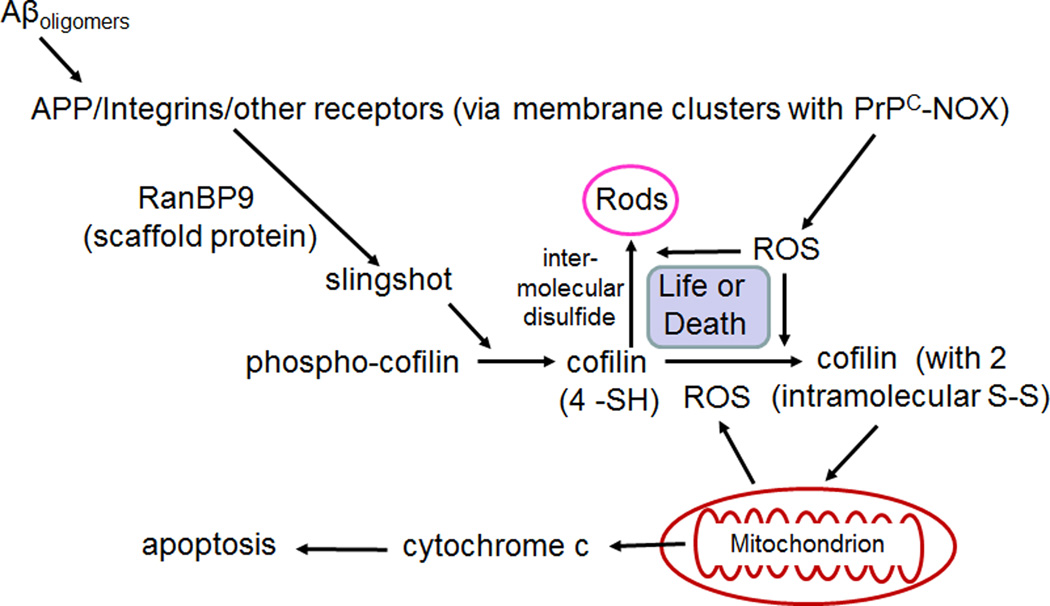
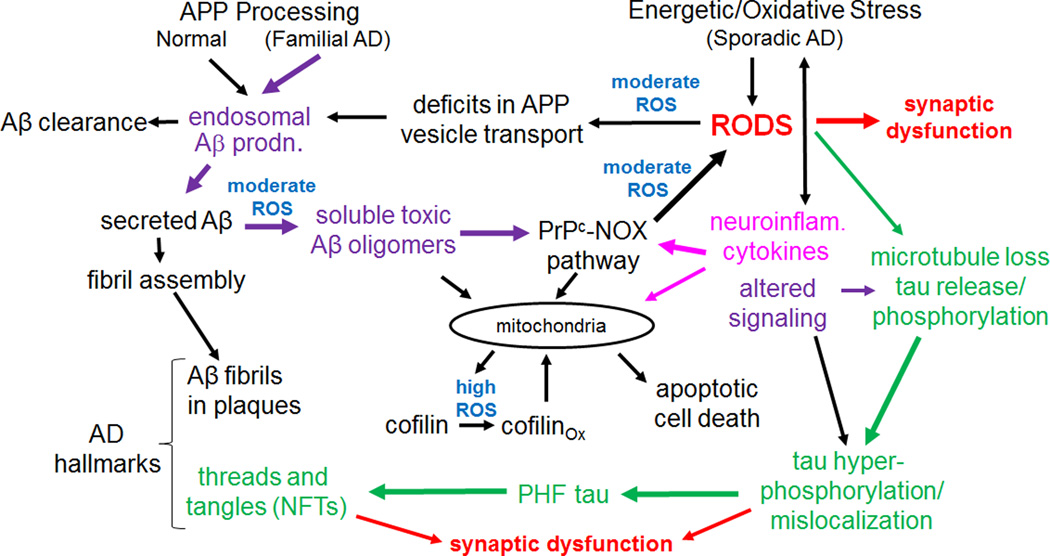
Cytoskeletal abnormalities and synaptic loss, typical of both familial and sporadic Alzheimer disease (AD), are induced by diverse stresses such as neuroinflammation, oxidative stress, and energetic stress, each of which may be initiated or enhanced by proinflammatory cytokines or amyloid-β (Aβ) peptides. Extracellular Aβ-containing plaques and intracellular phospho-tau-containing neurofibrillary tangles are postmortem pathologies required to confirm AD and have been the focus of most studies. However, AD brain, but not normal brain, also have increased levels of cytoplasmic rod-shaped bundles of filaments composed of ADF/cofilin-actin in a 1:1 complex (rods). Cofilin, the major ADF/cofilin isoform in mammalian neurons, severs actin filaments at low cofilin/actin ratios and stabilizes filaments at high cofilin/actin ratios. It binds cooperatively to ADP-actin subunits in F-actin. Cofilin is activated by dephosphorylation and may be oxidized in stressed neurons to form disulfide-linked dimers, required for bundling cofilin-actin filaments into stable rods. Rods form within neurites causing synaptic dysfunction by sequestering cofilin, disrupting normal actin dynamics, blocking transport, and exacerbating mitochondrial membrane potential loss. Aβ and proinflammatory cytokines induce rods through a cellular prion protein-dependent activation of NADPH oxidase and production of reactive oxygen species. Here we review recent advances in our understanding of cofilin biochemistry, rod formation, and the development of cognitive deficits. We will then discuss rod formation as a molecular pathway for synapse loss that may be common between all three prominent current AD hypotheses, thus making rods an attractive therapeutic target. © 2016 Wiley Periodicals, Inc.
Keywords: NADPH oxidase; amyloid-β; oxidative stress; prion signaling; proinflammatory cytokines.
© 2016 Wiley Periodicals, Inc.
Conflict of interest statement
The authors have no conflict of interest to declare.
Figures


References
-
- Agnew BJ, Minamide LS, Bamburg JR. Reactivation of phosphorylated actin depolymerizing factor and identification of the regulatory site. J Biol Chem. 1995;270:17582–17587. - PubMed
-
- Alzheimer’s Association. 2015 Alzheimer’s disease facts and figures. Alzheimer’s & Dementia. 2015;11:332–384. - PubMed
-
- Andrianantoandro E, Pollard TD. Mechanism of actin filament turnover by severing and nucleation at different concentrations of ADF/cofilin. Mol Cell. 2006;24:13–23. - PubMed
-
- Andziak B, Buffenstein R. Disparate patterns of age-related changes in lipid peroxidation in long-lived naked mole-rats and shorter lived mice. Aging Cell. 2006;5:525–532. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
