

SplAdder: identification, quantification and testing of alternative splicing events from RNA-Seq data
- PMID: 26873928
- PMCID: PMC4908322
- DOI: 10.1093/bioinformatics/btw076
SplAdder: identification, quantification and testing of alternative splicing events from RNA-Seq data
Abstract
Motivation: Understanding the occurrence and regulation of alternative splicing (AS) is a key task towards explaining the regulatory processes that shape the complex transcriptomes of higher eukaryotes. With the advent of high-throughput sequencing of RNA (RNA-Seq), the diversity of AS transcripts could be measured at an unprecedented depth. Although the catalog of known AS events has grown ever since, novel transcripts are commonly observed when working with less well annotated organisms, in the context of disease, or within large populations. Whereas an identification of complete transcripts is technically challenging and computationally expensive, focusing on single splicing events as a proxy for transcriptome characteristics is fruitful and sufficient for a wide range of analyses.
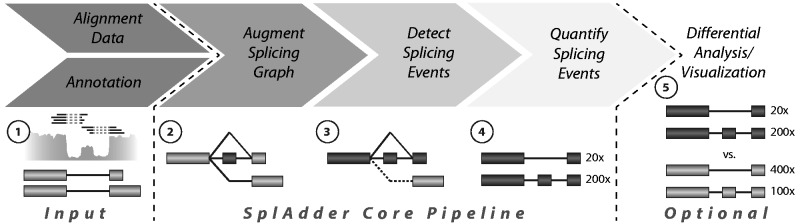
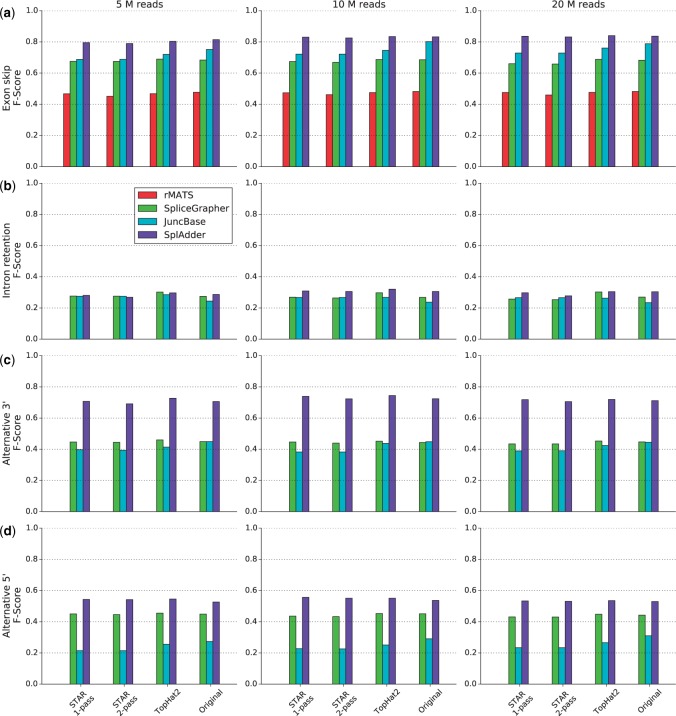
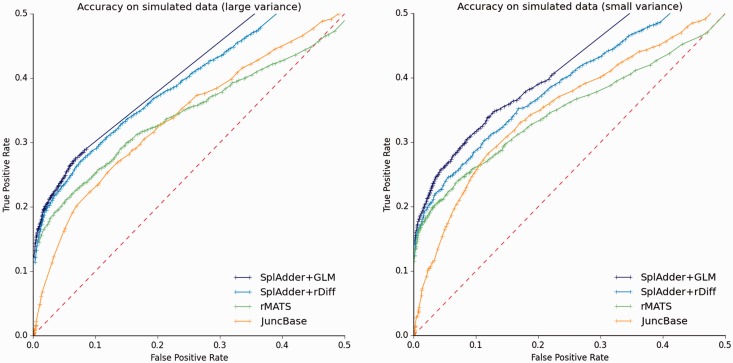
Results: We present SplAdder, an alternative splicing toolbox, that takes RNA-Seq alignments and an annotation file as input to (i) augment the annotation based on RNA-Seq evidence, (ii) identify alternative splicing events present in the augmented annotation graph, (iii) quantify and confirm these events based on the RNA-Seq data and (iv) test for significant quantitative differences between samples. Thereby, our main focus lies on performance, accuracy and usability.
Availability: Source code and documentation are available for download at http://github.com/ratschlab/spladder Example data, introductory information and a small tutorial are accessible via http://bioweb.me/spladder
Contacts: : andre.kahles@ratschlab.org or gunnar.ratsch@ratschlab.org
Supplementary information: Supplementary data are available at Bioinformatics online.
© The Author 2016. Published by Oxford University Press.
Figures
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
