

Improved Ribosome-Footprint and mRNA Measurements Provide Insights into Dynamics and Regulation of Yeast Translation
- PMID: 26876183
- PMCID: PMC4767672
- DOI: 10.1016/j.celrep.2016.01.043
Improved Ribosome-Footprint and mRNA Measurements Provide Insights into Dynamics and Regulation of Yeast Translation
Abstract
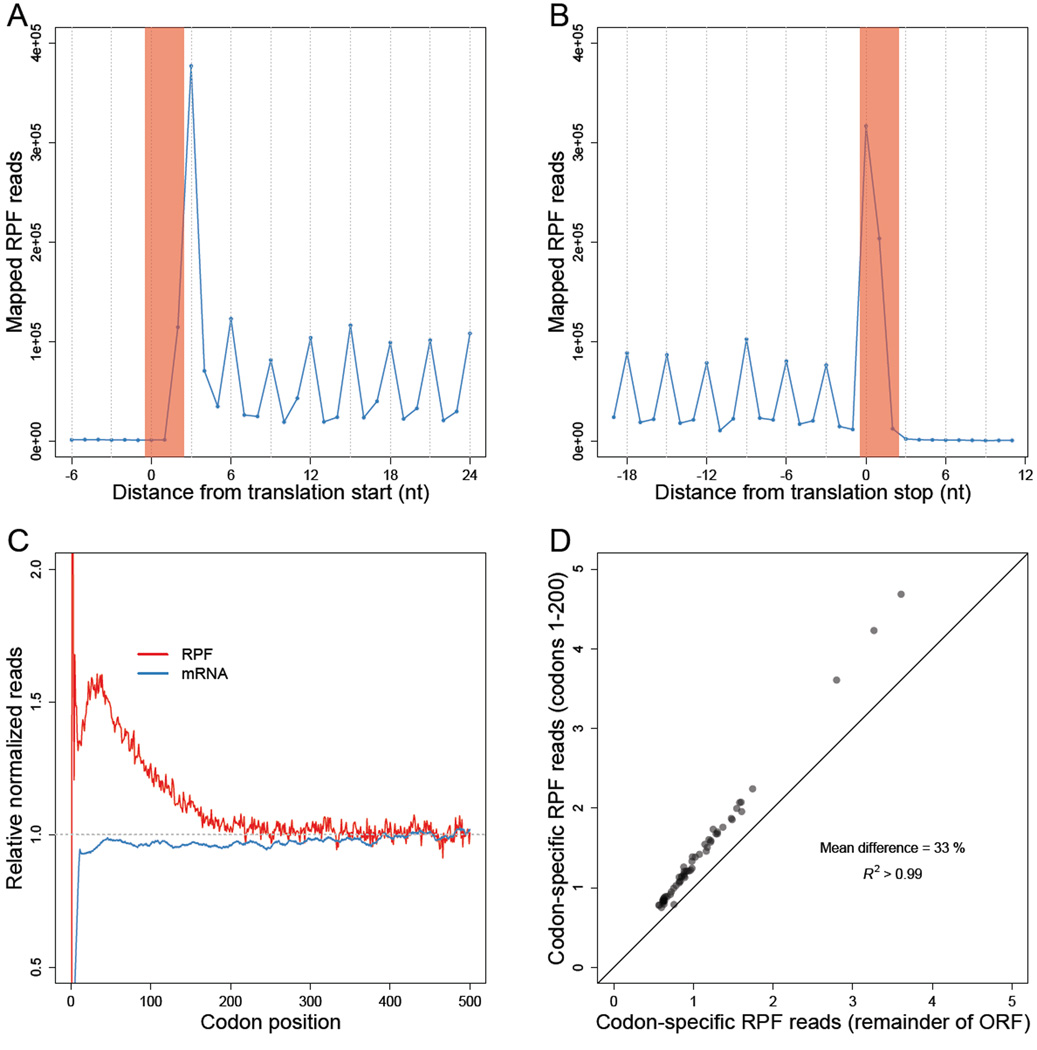
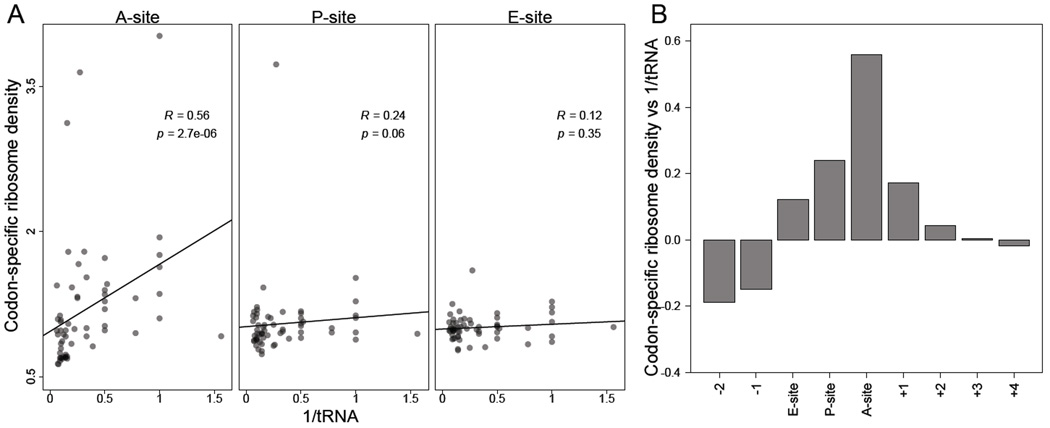
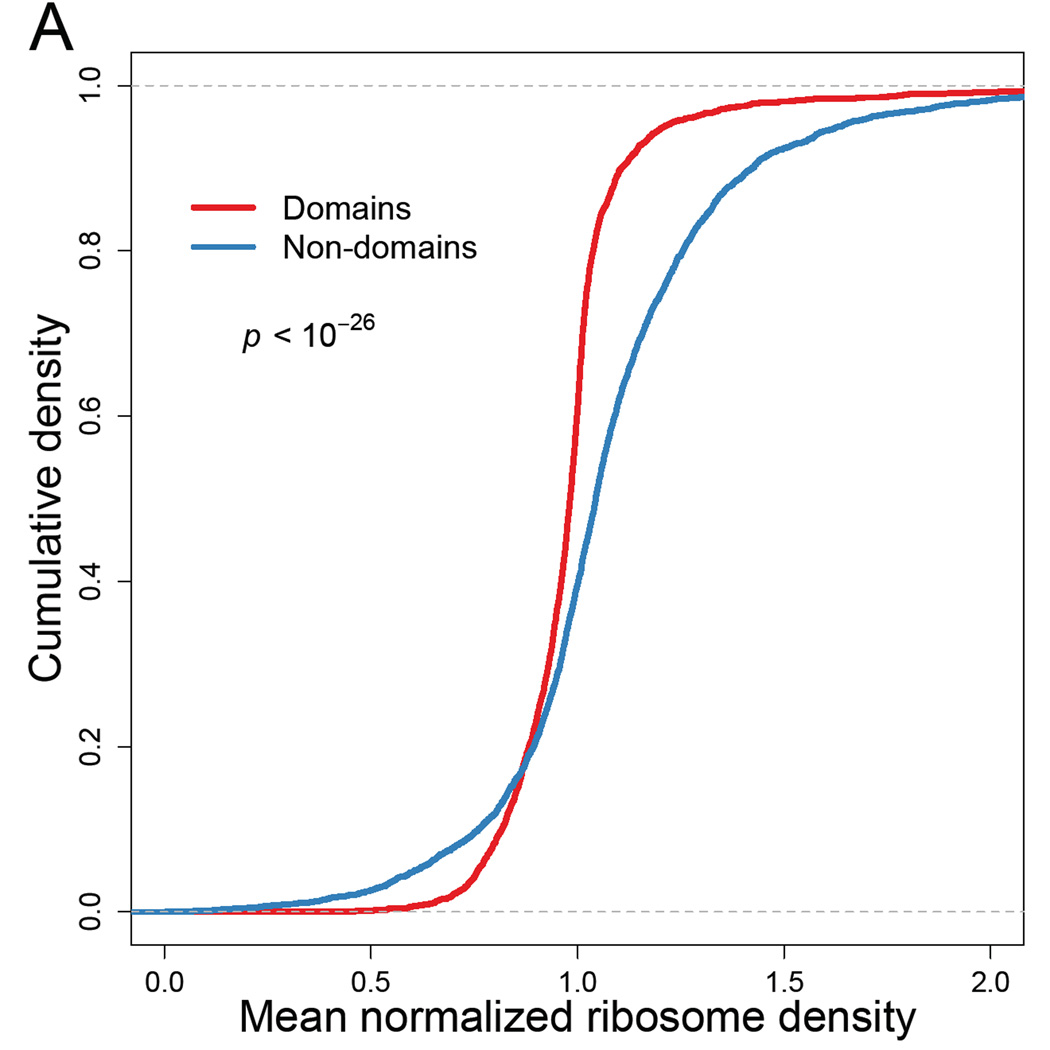
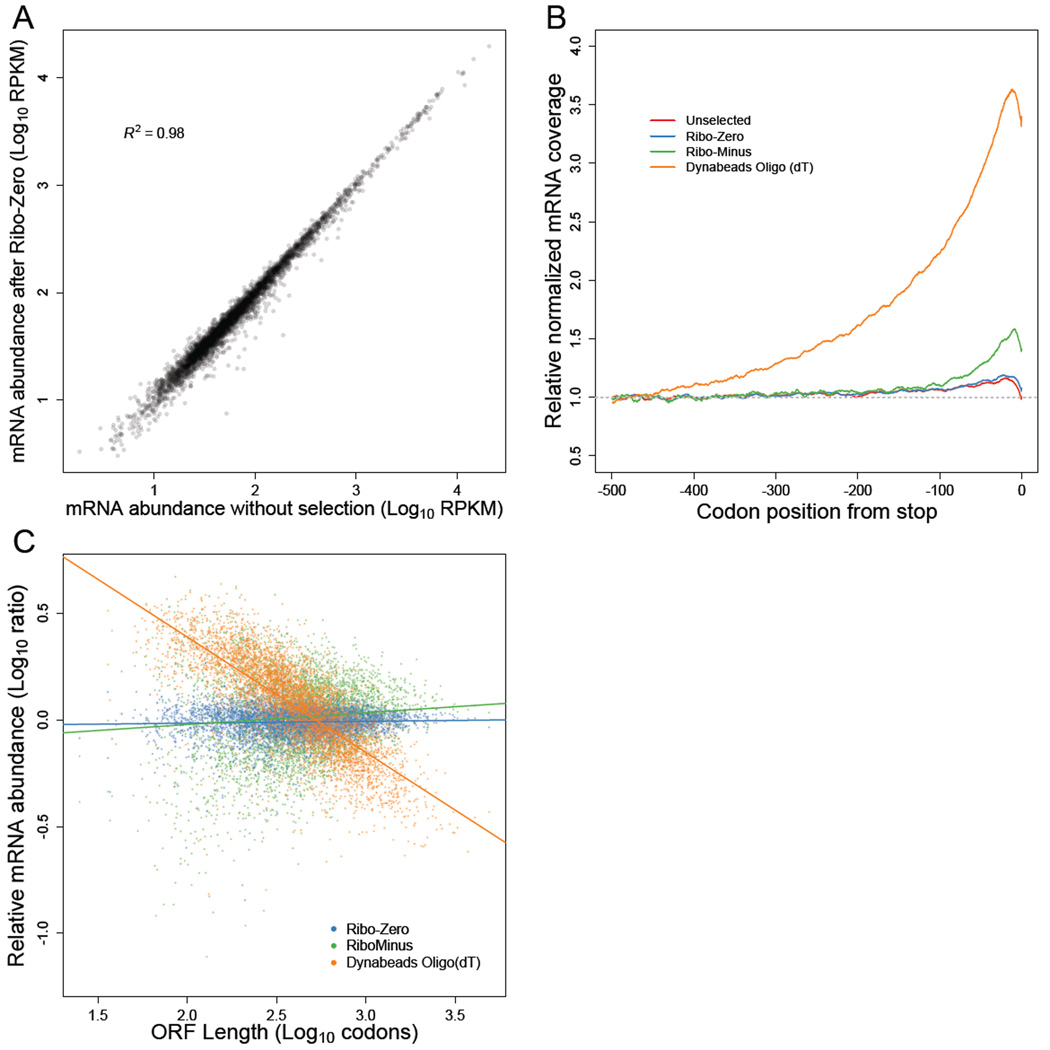
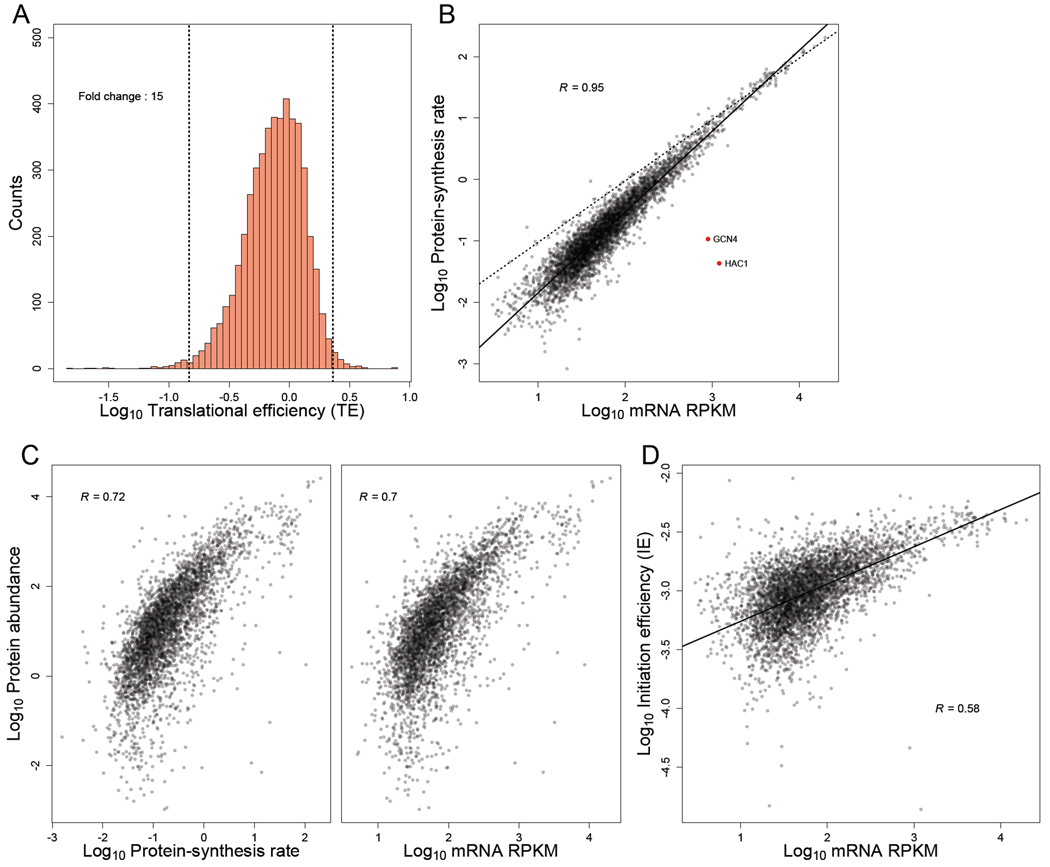
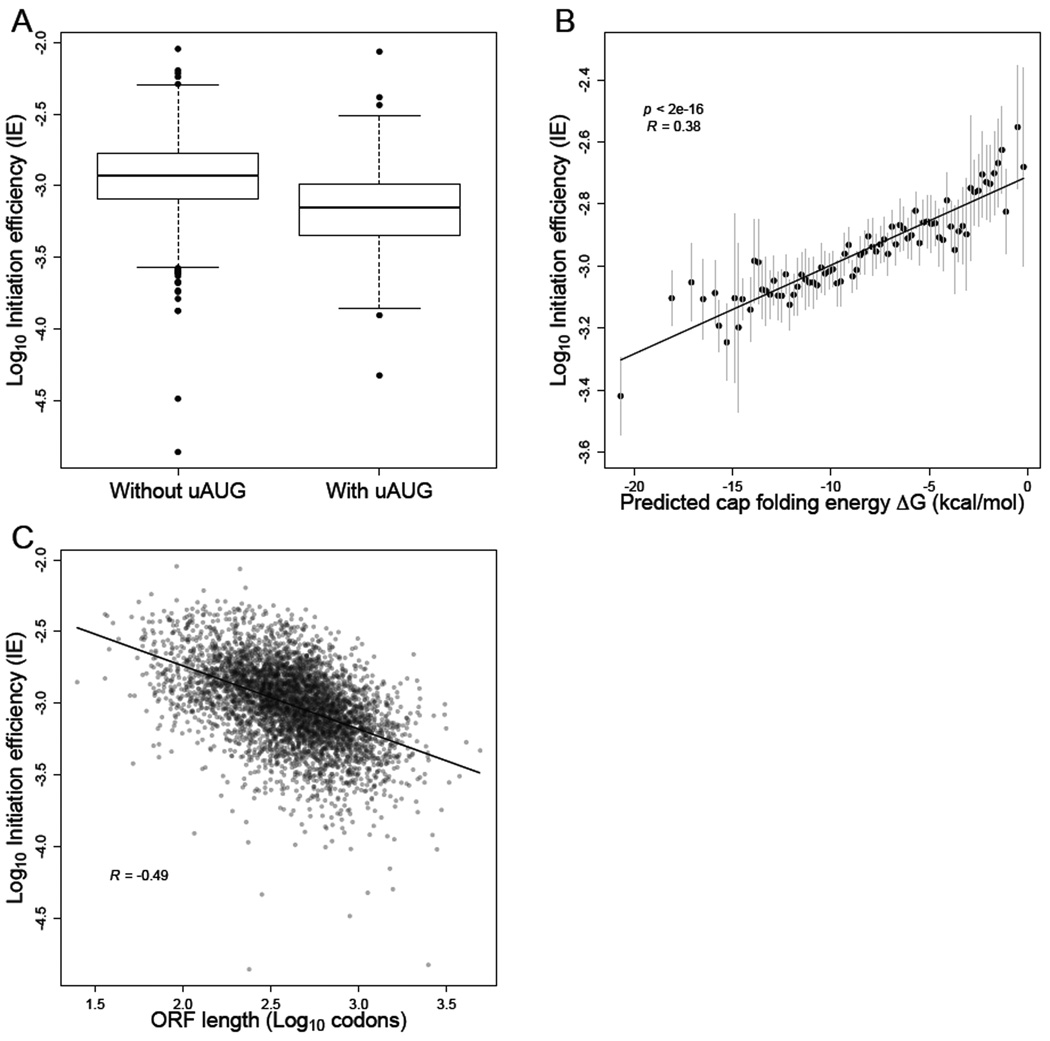
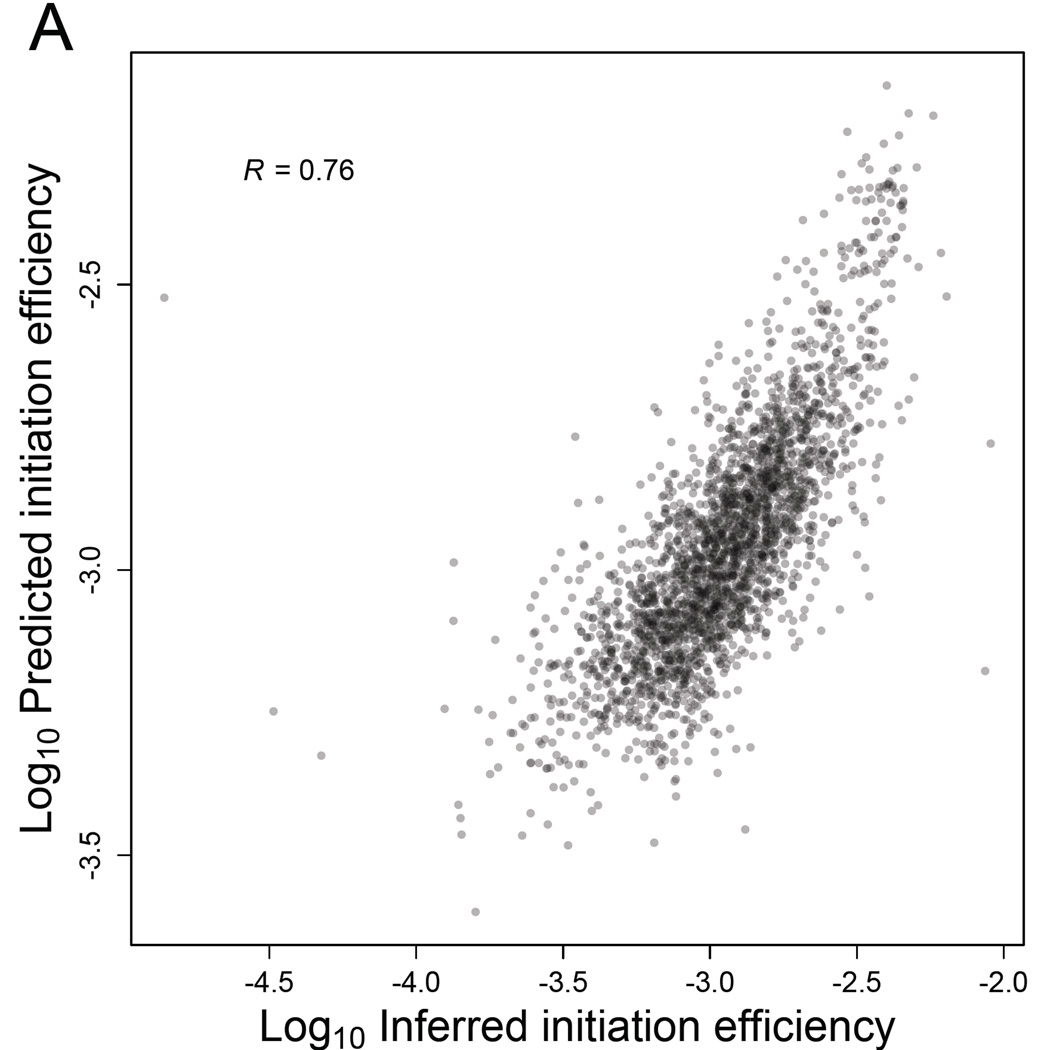
Ribosome-footprint profiling provides genome-wide snapshots of translation, but technical challenges can confound its analysis. Here, we use improved methods to obtain ribosome-footprint profiles and mRNA abundances that more faithfully reflect gene expression in Saccharomyces cerevisiae. Our results support proposals that both the beginning of coding regions and codons matching rare tRNAs are more slowly translated. They also indicate that emergent polypeptides with as few as three basic residues within a ten-residue window tend to slow translation. With the improved mRNA measurements, the variation attributable to translational control in exponentially growing yeast was less than previously reported, and most of this variation could be predicted with a simple model that considered mRNA abundance, upstream open reading frames, cap-proximal structure and nucleotide composition, and lengths of the coding and 5' UTRs. Collectively, our results provide a framework for executing and interpreting ribosome-profiling studies and reveal key features of translational control in yeast.
Copyright © 2016 The Authors. Published by Elsevier Inc. All rights reserved.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
