

Disruption of insulin receptor function inhibits proliferation in endocrine-resistant breast cancer cells
- PMID: 26876199
- PMCID: PMC4982805
- DOI: 10.1038/onc.2015.488
Disruption of insulin receptor function inhibits proliferation in endocrine-resistant breast cancer cells
Abstract
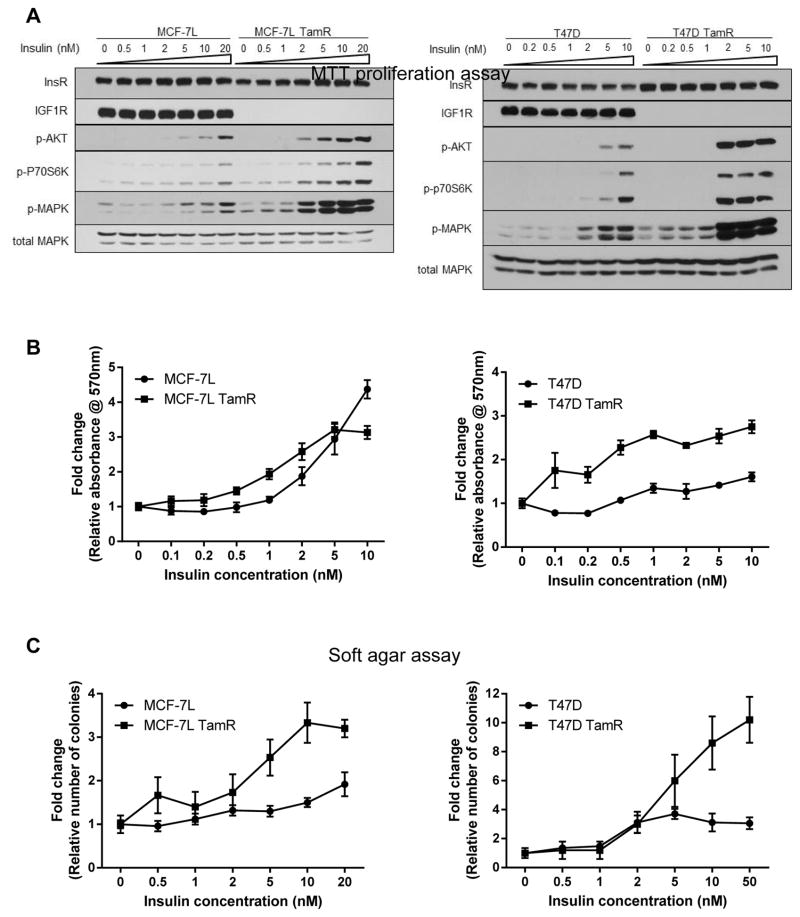
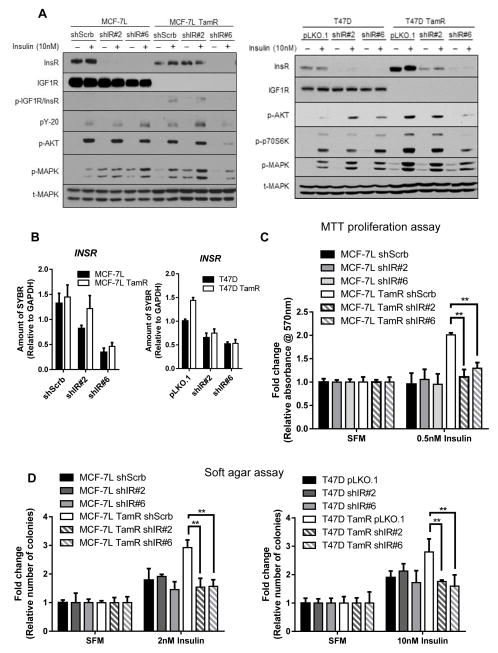
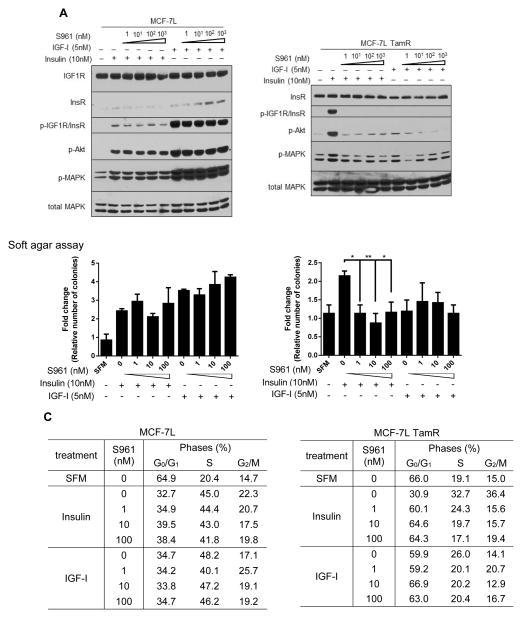
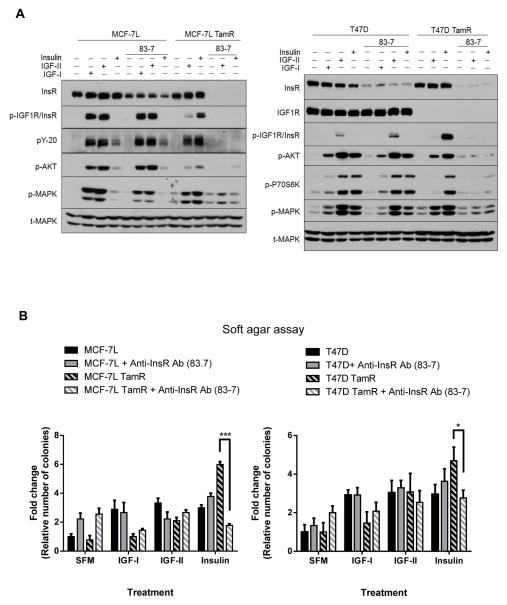
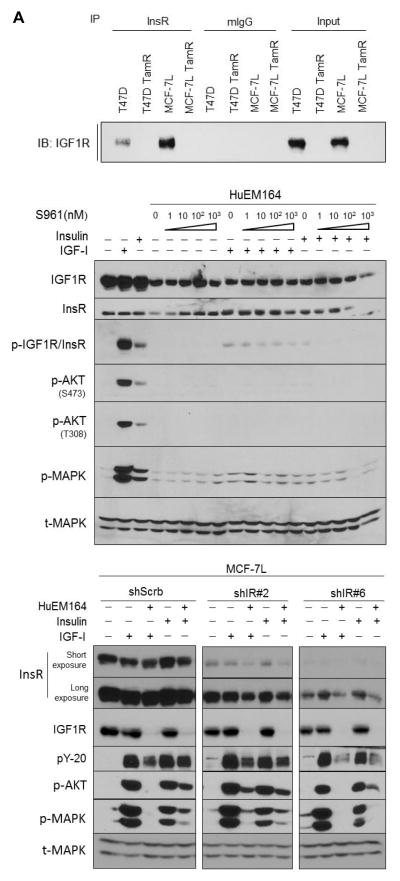
The insulin-like growth factor (IGF) system is a well-studied growth regulatory pathway implicated in breast cancer biology. Clinical trials testing monoclonal antibodies directed against the type I IGF receptor (IGF1R) in combination with estrogen receptor-α (ER) targeting have been completed, but failed to show benefits in patients with endocrine-resistant tumors compared to ER targeting alone. We have previously shown that the closely related insulin receptor (InsR) is expressed in tamoxifen-resistant (TamR) breast cancer cells. Here we examined if inhibition of InsR affected TamR breast cancer cells. InsR function was inhibited by three different mechanisms: InsR short hairpin RNA, a small InsR-blocking peptide, S961 and an InsR monoclonal antibody (mAb). Suppression of InsR function by these methods in TamR cells successfully blocked insulin-mediated signaling, monolayer proliferation, cell cycle progression and anchorage-independent growth. This strategy was not effective in parental cells likely because of the presence of IGFR /InsR hybrid receptors. Downregulation of IGF1R in conjunction with InsR inhibition was more effective in blocking IGF- and insulin-mediated signaling and growth in parental cells compared with single-receptor targeting alone. Our findings show TamR cells were stimulated by InsR and were not sensitive to IGF1R inhibition, whereas in tamoxifen-sensitive parental cancer cells, the presence of both receptors, especially hybrid receptors, allowed cross-reactivity of ligand-mediated activation and growth. To suppress the IGF system, targeting of both IGF1R and InsR is optimal in endocrine-sensitive and -resistant breast cancer.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures
Similar articles
-
Targeting Insulin Receptor in Breast Cancer Using Small Engineered Protein Scaffolds.Mol Cancer Ther. 2017 Jul;16(7):1324-1334. doi: 10.1158/1535-7163.MCT-16-0685. Epub 2017 May 3. Mol Cancer Ther. 2017. PMID: 28468775 Free PMC article.
-
Autocrine IGF-I/insulin receptor axis compensates for inhibition of AKT in ER-positive breast cancer cells with resistance to estrogen deprivation.Breast Cancer Res. 2013;15(4):R55. doi: 10.1186/bcr3449. Breast Cancer Res. 2013. PMID: 23844554 Free PMC article.
-
Controlled dimerization of insulin-like growth factor-1 and insulin receptors reveals shared and distinct activities of holo and hybrid receptors.J Biol Chem. 2018 Mar 9;293(10):3700-3709. doi: 10.1074/jbc.M117.789503. Epub 2018 Jan 12. J Biol Chem. 2018. PMID: 29330302 Free PMC article.
-
Inhibition of erbB receptor (HER) tyrosine kinases as a strategy to abrogate antiestrogen resistance in human breast cancer.Clin Cancer Res. 2001 Dec;7(12 Suppl):4436s-4442s; discussion 4411s-4412s. Clin Cancer Res. 2001. PMID: 11916237 Review.
-
Insulin and Insulin Receptors in Adipose Tissue Development.Int J Mol Sci. 2019 Feb 11;20(3):759. doi: 10.3390/ijms20030759. Int J Mol Sci. 2019. PMID: 30754657 Free PMC article. Review.
Cited by
-
Obesity and endocrine therapy resistance in breast cancer: Mechanistic insights and perspectives.Obes Rev. 2022 Feb;23(2):e13358. doi: 10.1111/obr.13358. Epub 2021 Sep 24. Obes Rev. 2022. PMID: 34559450 Free PMC article. Review.
-
S961, a biosynthetic insulin receptor antagonist, downregulates insulin receptor expression & suppresses the growth of breast cancer cells.Indian J Med Res. 2018 Jun;147(6):545-551. doi: 10.4103/ijmr.IJMR_403_17. Indian J Med Res. 2018. PMID: 30168485 Free PMC article.
-
IGF-1 Stimulates Glycolytic ATP Production in MCF-7L Cells.Int J Mol Sci. 2023 Jun 16;24(12):10209. doi: 10.3390/ijms241210209. Int J Mol Sci. 2023. PMID: 37373357 Free PMC article.
-
Insulin acts as a repressive factor to inhibit the ability of PAR2 to induce islet cell transdifferentiation.Islets. 2018 Sep 19;10(6):201-212. doi: 10.1080/19382014.2018.1472839. Islets. 2018. PMID: 29723131 Free PMC article.
-
Insulin Receptor Isoforms in Physiology and Disease: An Updated View.Endocr Rev. 2017 Oct 1;38(5):379-431. doi: 10.1210/er.2017-00073. Endocr Rev. 2017. PMID: 28973479 Free PMC article. Review.
References
-
- Nadji M, Gomez-Fernandez C, Ganjei-Azar P, Morales AR. Immunohistochemistry of estrogen and progesterone receptors reconsidered: experience with 5,993 breast cancers. Am J Clin Pathol. 2005;123:21–7. - PubMed
-
- Macaskill EJ, Renshaw L, Dixon JM. Neoadjuvant use of hormonal therapy in elderly patients with early or locally advanced hormone receptor-positive breast cancer. Oncologist. 2006;11:1081–8. - PubMed
-
- Paridaens RJ, Dirix LY, Beex LV, Nooij M, Cameron DA, Cufer T, et al. Phase III study comparing exemestane with tamoxifen as first-line hormonal treatment of metastatic breast cancer in postmenopausal women: the European Organisation for Research and Treatment of Cancer Breast Cancer Cooperative Group. J Clin Oncol. 2008;26:4883–90. - PMC - PubMed
-
- Ring A, Dowsett M. Mechanisms of tamoxifen resistance. Endocrine-related cancer. 2004;11:643–58. - PubMed
-
- Giuliano M, Schifp R, Osborne CK, Trivedi MV. Biological mechanisms and clinical implications of endocrine resistance in breast cancer. Breast. 2011;20(Suppl 3):S42–9. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
